Навигация
Организация наследственного аппарата клеток человека (уровни организации: генный, хромосомный, геномный)
37903
знака
3
таблицы
6
изображений
3. Организация наследственного аппарата клеток человека (уровни организации: генный, хромосомный, геномный).
Ген – участок ДНК, кодирующий синтез одной полипептидной цепи аминокислот (одной молекулы белка) размеры гена определяются числом пар нуклеотидов. Есть гены размером в 59 пар нуклеотидов (п. н.) – у фага Т-4, 4 – в несколько тысяч п. н. (большинство генов человека). Учёные считают, что в генотипе человека насчитывается около 1 миллиона генов.
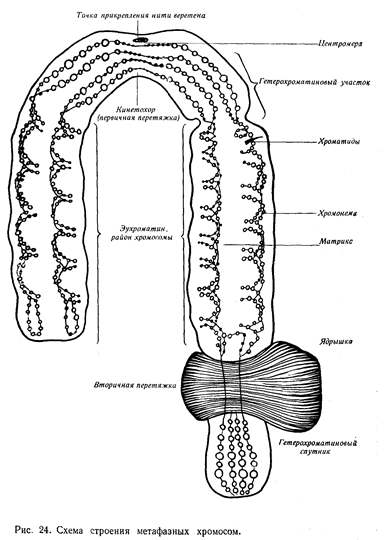
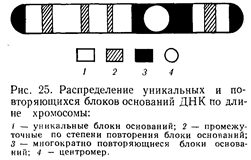
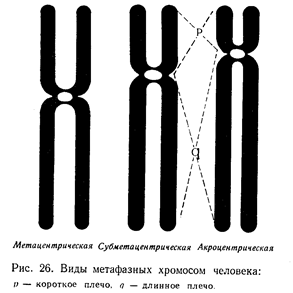
Хромосома - (в переводе – «окрашенное тельце») сложное образование внутри ядра, состоит из: ДНК, белков, РНК, липидов, углеводов. В одной хромосоме размещается (локализуется) много генов. Хромосомы имеют разную форму. Форма хромосомы определяется положением центромеры (первичной перетяжки, к которой присоединяются нити веретена деления в митозе). Если центромера делит хромосому пополам, то у неё образуются равные плечи, поэтому такую хромосому называют «равноплечей» или метацентрической.
Если центромера немножко смещена в сторону одного плеча – это «неравноплечая» или субметацентрическая хромосома.
Если центромера делит хромосому так, что одно плечо короче другого на 75%, то её называют «резко неравноплечая» или – акроцентрическая.
Если же центромера располагается в одном конце хромосомы, то хромосому называют телоцентрической.
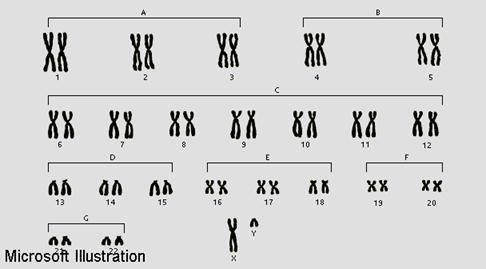
Совокупность хромосом ядра, их число, форма и структура называется кариотипом. У человека кариотип 2n=46 был установлен в 1956г. двумя учёными: Дж. Тийо и А. Леваном. Кариотип человека изображают в виде идеограммы – схемы, на которой хромосомы располагают в ряд по мере убывания их длинны, и по одной из каждой пары. Все хромосомы объединены в 7 групп, обозначаемых буквами римского алфавита. Распределены хромосомы на идеограмме с учётом размеров хромосом и локализации центромерного участка, и каждая хромосома имеет свой номер (арабская цифра).
Группа А – 1 2 3
Группа В – 4 5
Группа С – 6 7 8 9 10 11 12
Группа D – 13 14 15
Группа Е – 16 17 18
Группа F – 19 20
Группа G – 21 22 половые хромосомы Х y (23)
В кариотипе мужчин и женщин есть одинаковые хромосомы, их большинство – 44 – это неполовые хромосомы или аутосомы (44А); и есть одна пара хромосом (23), по которой отмечается различие: у женщин ХХ, у мужчин Ху.
Если признак контролируется доминантным геном, локализованным в какой-либо аутосоме, то его называют аутосомно-доминантный; а рецессивным геном – аутосомно-рецессивным. Наследование признаков, контролируемых генами аутосом, подчиняется законам Менделя. Менделирующих признаков, в том числе и болезней, у человека около 3 тыс.
| Тип наследования. | 1978 год. |
| Аутосомно-доминантный | 1489 |
| Аутосомно-рецессивный | 1117 |
| Сцепленный с Х-хромосомой | 205 |
| Всего… | 2811 |
Если признак контролируется генами, локализованными в Х-хромосоме, он называется сцепленным с полом (или с Х-хромосомой). Если обнаруживается сцепление с У-хромосомой, то признак называют голандрическим. Признак, сцепленный с Х-хромосомой подчиняется правилу «крисс-кросса» (крест-накрест): от матери – сыну, от отца к дочери. Голандрический признак передаётся от отца – сыну, т. е. Только по мужской линии.
Геном - совокупность гаплоидного (1п) набора хромосом (23 хромосомы).
4. Мутационный процесс и наследственные заболевания человека:
а) механизм генных мутаций. Болезни обмена веществ и молекулярные болезни человека. Наследование генных аномалий.
Мутации происходят на каждом из перечисленных уровней, и их называют генными, хромосомными, геномными.
Многие мутации являются причиной наследственных заболеваний, которых насчитывается около 2000. Изучение и возможное предотвращение последствий генетических дефектов человека – предмет медицинской генетики. Это так называемый «генетический груз» популяций людей.
Рассмотрим роль генных мутаций в формировании наследственных заболеваний.
Генные мутации называют ещё точковыми мутациями. Они обусловлены изменением молекулярной структуры ДНК. В соответствующем участке ДНК эти изменения касаются нуклеотидов, входящих в состав гена. Такие изменения нуклеотидного состава гена могут быть 4-х типов:
1. Вставка нового нуклеотида
2. Выпадение нуклеотида
3. Перестановка положения нуклеотидов
4. Замена нуклеотидов.
Любое из перечисленных изменений приводит к изменению триплета (триплетов) в И-РНК, а это влечёт за собой изменение состава аминокислот в полипептиде, т.е. приводит к нарушению синтеза нормальной молекулы белка. Например:
Много сведений об изменении гена дало исследование гемоглобина. Было установлено, что при тяжёлом заболевании – серповидноклеточной анемии – эритроциты содержат аномальный гемоглобин (HbS) и имеют необычную, отличающуюся от нормальной форму. Нормальный гемоглобин (HbA)содержит четыре полипептидные цепи (две так называемые α- и две β-цепи, а α-цепи HbS не отличаются от α-цепей HbA) Различие HbA и S заключается лишь в замене одного аминокислотного остатка, а именно глютаминовой кислоты, на валин в шестом положении β-цепи.
Последовательность аминокислот в начальном участке β-цепи нормального (HbA) изменённого (HbS) гемоглобина следующая:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| HbA | Вал... | Гис… | Лей... | Тре… | Про... | Глю... | Глю... | Лиз... |
| HbS | Вал... | Гис… | Лей... | Тре… | Про... | Вал... | Вал... | Лиз... |
Глютамированную кислоту кодирует в мРНК триплет ГАГ. Изменения в мРНК, ответственное за включение валина вместо глютаминовой кислоты, состоит в замене одного нуклеотида, а именно А на У, вследствие чего получается триплет ГУГ, кодирующий валин. На этом основании можно заключить, что в структурном гене ДНК, кодирующем β-цепь гемоглобина, семнадцатый нуклеотид, в норме представленный Т, заменён на А.
Наследственных болезней, вызванных генными мутациями, насчитывается около 1500. Их условно подразделяют на: болезни обмена веществ и молекулярные болезни.
Болезней обмена веществ насчитывается около 600, они затрагивают изменения аминокислотного, углеводного и липидного состава клетки. Некоторые мутации вызывают возникновение даже злокачественных образований.
| Признак | Характер | наследования |
| Доминантный | рецессивный | |
| Обмен веществ: аминокислотный углеводный липидный Злокачественные заболевания | Нейрофиброматоз | Альбинизм Фенилкетонурия Галактоземия Мукополисахаридозы (гаргонтилизм) Амавротическая семейная идиотия (болезнь Тея-Сакса) Глиома сетчатки глаза Врождённый ихтиоз |
Из этой таблицы явствует, что генные заболевания могут наследоваться как по аутосомно-доминантному, так и по аутосомно-рецессивному типу.
По доминантному типу передаётся нейрофиброматоз, – хроническое заболевание, характеризующееся множественным образованием опухолей нервных стволов. Такие опухоли могут локализоваться в любых органах и тканях ( в том числе и в ЦНС), но чаще всего они встречаются на коже, где имеют вид пигментированных бородавок с избыточным ростом волос. К симптомам заболевания относится даже отставание физического и умственного развития.
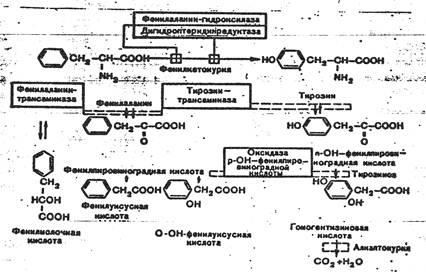
По рецессивному признаку передаётся фенилкетонурия (болезнь Феллинга) – резкое повышение содержания в крови и ликворе аминокислоты фенилатина и превращение её в ряд продуктов, например в фенилпировиноградную и фенилмолочную кислоты. В отличие от гомогентезиновой кислоты, которая не оказывает явного неблагоприятного влияния на ткани мозга, продукты, образующиеся при фенилкетонурии, оказываются крайне токсичными. Поэтому у детей при этой патологии наблюдается резко выраженная умственная отсталость. Заболевание выражается также в снижении количества пигмента меланина, поэтому больные всегда выглядят, как голубоглазые блондины со светлой кожей. В настоящее время диагноз можно поставить при рождении ребёнка экспресс-методом: на смоченную мочой плёнку наносят 5 капель 10% раствора FeCl3 или добавляют в 1мл подкислённой мочи (при заболевании наблюдается быстро проходящее потемнение).
Галактоземия – нарушение углеводного обмена. Она обусловлена нарушением деятельности печени, накоплением в тканях (в том числе и крови) галактозы. Без лечения развивается цирроз печени; в патологический процесс вовлекаются и другие жизненно важные органы. В конечном итоге болезнь приводит к слабоумию и ранней смерти. В начале жизни, как только новорождённый начинает получать молоко, наблюдается желтуха, рвота, диспепсические расстройства, падение массы тела. При ранней диагностике детей до трёхлетнего возраста переводят на безмолочное вскармливание, т. е. исключают продукты, содержащие галактозу. Такие дети развиваются нормально и отклонений в психике у них не наблюдается. Носительство гена, вызывающего заболевание, т. е. число гетерозигот, составляет в среднем 1:70 000.
Аномалии, связанные с нарушениями распада некоторых углеводосодержащих соединений, вызывают развитие мукополисахаридозов (гаргоилизмы). При этих заболеваниях поражена соединительная ткань, а следовательно, страдают опорно-трофические функции и моторика. Доя больных мукополисахаридозом характерно уродливое телосложение (дети напоминают уродцев – гаргоидов), наличие множественных пороков внутренних органов ( печени органов , сердца, аорты, нервной системы) и глаз.
Нарушение липидного обмена – амавротическая идиотия (болезнь Тея-Сакса), связанная с отсутствием фермента гексосаминдазы А – тяжёлое расстройство нервной системы. Эту болезнь можно обнаружить лишь во второй половине первого года жизни ребёнка, когда наблюдается прогрессирующее отставание физического развития, нарушение зрения и интеллекта. В дальнейшем больной слепнет, развивается слабоумие и полная беспомощность. Тяжёлые симптомы нарастают, что приводит к смерти ребёнка до 4 – 5 лет.
Молекулярные болезни лучше всего изучены на элементах крови. Известно около 50 наследственных болезней крови. Некоторые из них наследуются по типу неполного доминирования. Например два вида гемоглобингопатий: серповидноклеточная анемия и талассимия (болезнь Кули). Гемоглобинопатии выражаются в гемолизе – в распаде аномальных эритроцитов. При этом наблюдается кислородное голодание, приступы лихорадки колики типа желчнокаменных и др. симптомы, которые могут закончиться смертью. Особенно тяжело эти заболевания протекают у гомозигот по данному признаку.
Ген серповидноклеточной анемии S, ответственный за синтез аномального гемоглобина HbS, приводит к образованию ненормальной серповидной формы эритроцитов. Этот ген очень часто встречается в Средиземноморье (в Греции), Центральной Африке, несколько реже в других частях африканского континента, В Юго-Восточной Азии - в Индии). Распространение этого гемоглобиноза совпадает с распространением тяжёлой формы тропической малярии и её возбудителя – кровяного споровика Plasmodium falciparum. Малярийные плазмодии способны развиваться лишь в нормальных эритроцитах. В ьторгн76серповидноклеточных эритроцитах гомозиготы они не развиваются совсем, поэтому и гетерозиготы , имеющие частично нормальные, частично серповидноклеточные эритроциты, либо не болеют, либо болеют в более лёгкой форме.
Другой ген – Т, также влияющий на свойства крови, в гомозиготном состоянии (ТТ) приводят к развитию иного, несколько легче протекающего гемоглобиноза – талассемии (микроцитарная форма анемии). Особенно распространена талассемия на побережье Средиземного моря ( Италия, Греция, Кипр), в Бирме, Бенгалии, а в России – в Средней Азии (обычно в кишлаках благодаря близкородственным бракам), в Азербайджане; отдельные очаги описаны в Узбекистане, у бухарских евреев.
Больные талассемией имеют характерный башенный череп, кости его деформированы и имеют вид «иголок ежа». Такие больные (ТТ) обычно не доживают до десятилетнего возраста, гетерозиготы же (Тт) практически мало чем отличаются от здоровых людей (тт).
Некоторые генные заболевания сцеплены с полом. Примером такого рода наследования является гемофилия, агаммаглобулинемия, несахарный диабет, дальтонизм и облысение.
В крови людей, страдающих гемофилией, нет компонента фибриногена, необходимого для её быстрого свёртывания. У таких людей происходит потеря большого количества крови даже при легких ранениях и незначительных операциях. Рассматривая историю рода, в котором есть ген, вызывающий гемофилию, учёные установили, что это заболевание передаётся потомству здоровыми женщинами, но не передаётся мужчинами. А подвержены ему только они. Когда поражённый мужчина женится на нормальной женщине, его дети и внуки от сыновей оказываются здоровыми. Среди его внуков от дочерей часть мальчиков страдает гемофилией, в то время как все девочки здоровы. Но некоторые из них имеют больных сыновей. Наследование гемофилии подчинено закономерности передачи рецессивного признака, сцеплённого с полом.
Другой широко распространённый у человека ген, сцеплённый с полом, вызывает цветовую слепоту. Этот ген рецессивен по отношению к нормальному. Мужчины, имеющие один ген дальтонизма, оказываются дальтониками, а женщины – потенциальными носителями. Это объясняет гораздо большую частоту дальтоников среди мужчин. Только в браке больных мужчин с женщинами, имеющими соответствующий ген, могут рождаться девочки-дальтоники.
б) хромосомные мутации, их разнообразие и проявление в форме синдромов.
Хромосомные болезни. Известно около 300 хромосомных синдромов, которые могут быть обусловлены изменением числа хромосом – аутосом (синдром Дауна) или половых хромосом (синдромы: Шерешевского – Тернера, Кляйнфельтера). Если обнаруживается одна лишняя хромосома (46+1), то это трисомия. Например синдром Дауна возникает при трисомии по 21 хромосоме (обозначают 21+).
Впервые открытие того, что синдромы врождённых пороков развития могут быть обусловлены отклонениями в составе хромосом, произошло в 1959 г. на болезни Дауна, клиническое описание которой было сделано ещё в прошлом веке. Открытие последовало за разработкой к концу 50-х годов эффективных методов определения числа и морфологии хромосом в клетках человека и млекопитающих.
Синдром Клайнфельтера – это группа клинически сходных отклонений в половом, соматическом и психическом развитии, которые развиваются у индивидуумов мужского пола при полных или частичных Х- или Y- полисомиях. Его суммарная частота 2,5 на 1000 живорожденных мальчиков.
Если одной хромосомы не хватает (46-1=45) – это моносомия. Если моносомия у женщин по половым хромосомам, то обозначают ХО.
Часты синдромы Шерешевского - Тернера (частота 0,7 на 1000 новорожденных девочек) и трипло-Х (1,4 на 1000 девочек). Клинические проявления синдрома в виде отставания в росте, отклонений в строении лица, шеи и др. проявляются в ранние годы, но основная симптоматика, выражающаяся в отсутствии развития или недоразвития вторичных половых признаков, в первичной аменорее, развивается в годы полового созревания. Взрослые пациенты бесплодны.
Наиболее частой из них и достаточно известной среди врачей и населения является трисомия по хромосоме 21, или болезнь Дауна.
На втором месте по частоте находится трисомия по хромосоме 18, или синдром Эдвардса. Она встречается в 10 раз реже болезни Дауна, пороки развития тяжелее; такие младенцы погибают в основном на первом году жизни.
Ещё реже, с частотой 7:100 000, рождаются живые дети с трисомией по хромосоме 13 (синдром Патау). Очень редки также трисомии по аутосомам 8 и 9.
Изменение числа половых хромосом оказывают менее вредное влияние на организм, чем аномалии аутосом. Большинство аутосомных хромосомных мутаций летально, в связи с чем эмбрион погибает на ранних сроках беременности.
Не только изменение числа хромосом, но и аномалии их структуры (делеции) вызывают хромосомные заболевания.
Синдромы, обусловленные делециями: 4р- (синдром Вольфа – Хиршхорна), 5р- (синдром кошачьего крика), 9p-, 13q-, 18q-, 18r, 21q-, 22q-.
В качестве примера хромосомных мутаций приведём 5p – утрата короткого плеча (p) 5-й хромосомы, или синдром «кошачьего крика» (название обусловлено сходством плача ребёнка с мяуканьем кошки). Такой крик объясняется не аномалией голосового аппарата, а нарушениями центральной нервной системы. Для синдрома 5p характерны микрогнаитя (от греческого гнатос – челюсть) и синдактилия, которые дополняют фенотипическую картину синдрома. У больных отмечается понижение сопротивляемости к инфекциям, поэтому относительно часто они умирают рано. Отягощающим фактором являются различные нарушения внутренних органов (аномалии сердца, почек, грыжи и др.).
Делеция – утеря участка хромосомы. Условное обозначение: 5р- (пятая хромосома, утрата в плече р).
в) геномные мутации и их последствия.
Геномные мутации – это полиплодия – у человека редкое явление. Описаны редкие триплоиды и тетраплоиды в основном среди спонтанно абортированных эмбрионов или плодов и среди мертворождений. Новорождённые с такими нарушениями живут несколько дней.
5.Факторы, вызывающие мутации наследственного аппарата.
Факторы вызывающие возникновение мутаций. Факторами, вызывающими (индуцирующими) мутации, могут быть самые разнообразные влияния внешней среды: температура, ультрафиолетовое излучение, радиация (как естественная, так и искусственная), действия различных химических соединений – мутагенов. Мутагенами называют агенты внешней среды, вызывающие те или иные изменения генотипа – мутацию, а сам процесс образования мутаций – мутагенезом.
Радиоактивным мутагенезом начали заниматься в 20-х годах нашего столетия. В 1925 г. советские учёные Г. С. Филиппов и Г. А. Надсон впервые в истории генетики применили рентгеновские лучи для получения мутаций у дрожжей. Через год американский исследователь Г. Меллер (в последствии дважды лауреат Нобелевской премии), длительное время работавший в Москве, в институте, руководимом Н. К. Кольцовым, применил тот же мутаген на дрозофиле.
Химический мутагенез впервые целенаправленно начали изучать сотрудник Н. К. Кольцова В. В. Сахаров в 1931 г. на дрозофиле при воздействии на её яйца йодом, а позже М. Е. Лобашов.
К химическим мутагенам относятся самые разнообразные вещества (алкилирующие соединения, перекись водорода, альдегиды и кетоны, азотная кислота и её аналоги, различные антиметаболиты, соли тяжёлых металлов, красители, обладающие основными свойствами, вещества ароматического ряда), инсектициды (от лат. insecta – насекомые, cida – убийца), гербициды (то лат. herba – трава), наркотики, алкоголь, никотин, некоторые лекарственные вещества и многие другие.
Генетически активные факторы можно разделить на 3 категории: физические, химические и биологические.
Физические факторы. К их числу относятся различные виды ионизирующей радиации и ультрафиолетовое излучение. Исследование действия радиации на мутационный процесс показало, что пороговая доза в этом случае отсутствует, и даже самые небольшие дозы повышают вероятность возникновения мутаций в популяции. Повышение частоты мутаций опасно не столько в индивидуальном плане, сколько с точки зрения увеличения генетического груза популяции. Например, облучение одного из супругов дозой в пределах удваивающей частоту мутаций (1,0 – 1,5 Гй) незначительно повышает опасность иметь больного ребёнка (с уровня 4 - 5% до уровня 5 – 6%). Если такую же дозу получит население целого района, то число наследственных заболеваний в популяции через поколение удвоится.
Химические факторы. Химизация сельского хозяйства и других областей человеческой деятельности, развитие химической промышленности обусловили синтез огромного потока веществ (в общей сумме от 3,5 до 4,3 млн.), в том числе таких, которых в биосфере никогда не было за миллионы лет предшествующей эволюции. Это означает прежде всего неразложимость и таким образом длительное сохранение чужеродных веществ попадающих в окружающую среду. То, что было принято первоначально за достижения в борьбе с вредными насекомыми, в дальнейшем обернулось сложной проблемой. Широкое применение в 40 – 60-е годы инсектицида ДДТ, относящегося к классу хлорированных углеводородов, привело к его распространению по всему земному шару вплоть до льдов Антарктиды.
Большинство пестицидов обладает большой устойчивостью к химическому и биологическому разложению и имеет высокий уровень токсичности.
Биологические факторы. Наряду с физическими и химическими мутагенами генетической активностью обладают также некоторые факторы биологической природы. Механизмы мутагенного эффекта этих факторов изучены наименее подробно. В конце 30-х годов С, М. Гершензоном начаты исследования мутагенеза у дрозофилы под действием экзогенной ДНК и вирусов. С тех пор установлен мутагенный эффект многих вирусных инфекций и для человека. Аберрации хромосом в соматических клетках вызывают вирусы оспы, кори, ветряной оспы, эпидемического паротита, гриппа, гепатита и др.
6. Значение диагностики и лечение от наследственных болезней.
По мере развития медицины возможность выявления наследственных заболеваний увеличивается. Этот фактор указывает на растущее значение медицинской генетики и генетики человека. Меры, принятые при раннем выявлении наследственных болезней, могут предотвратить их развитие. Диетологические меры позволяют избежать патологических последствий, например при галактоземии, фенилкетонурии и других наследственных болезнях обмена.
При диагностике наследственных заболеваний Н. П. Бочков с сотрудниками рекомендует руководствоваться следующим:
1. Применять клинико-генеалогический метод, который позволяет обнаруживать «семейные» болезни.
2. Часто к наследственным относятся заболевания, повторяющиеся хронически и длительно не поддающиеся лечению, особенно в детском возрасте.
3. На возможную наследственную форму заболевания указывают редко встречающиеся специфические симптомы.
4. То же относится к патологическим изменениям многих органов и систем.
Для многих наследственных заболеваний стала возможна так называемая пренатальная (т. е. до рождения) диагностика. Это метод амниоцентеза, который заключается в получении с помощью шприца 10-15 мл амниотической жидкости, в которой находятся клетки плода. Так определяют соотношение метаболитов, отражающих нормальное или патологическое состояние плода. Культивируемые эмбриональные клетки используют для определения числа хромосом и выявления возможных хромосомных аномалий.
Методы лечения:
Первый метод – диетотерапия: исключение или добавление определённых веществ в рацион. Примером могут служить диеты: при галактоземии, при фенилкетонурии, при гликогенозах и т. д.
Второй метод – возмещение не синтезируемых в организме веществ, так называемая заместительная терапия. При сахарном диабете используют инсулин. Известны и другие примеры заместительной терапии: введение антигемофильного глобулина при гемофилии, гамма-глобулина при иммунодефицитных состояниях и др.
Третий метод – удаление токсических продуктов обмена из организма. Характерным примером может служить выведение меди при гепатолентикулярной дегенерации с помощью пеницилламина, сульфида калия и других препаратов.
Четвёртый метод – медиеометозное воздействие, основная задача которого оказать влияние на механизмы синтеза ферментов. Например, назначение барбитуратов при болезни Криглера – Найара способствует индукции синтеза фермента глюкоронил-трансферазы. Витамин В6 активизирует фермент цистатионинсинтетазу и обладает лечебным действием при гомоцистинурии.
Пятый метод – исключение из употребления лекарств, как, например, барбитуратов при порфирии, сульфаниламидов при глюкозо-6-фосфатдегидрогеназы.
Шестой метод – хирургическое лечение. Прежде всего это относится к новым методам пластической и восстановительной хирургии (врождённые пороки сердца и сосудов, расщепление губы и нёба, различные костные дефекты и деформации).
Медико-генетичесое консультирование – это сложный процесс, требующий от консультанта всесторонней подготовки по генетике и по теории вероятности, так как он сталкивается с решением многих разнообразных генетических задач. Кроме того, консультант должен иметь опыт в области клинической медицины и хорошо знать наследственную патологию в связи с необходимостью уточнять диагноз наследственного заболевания. Наконец, консультант должен быть высокогуманен и принципиален в отношении позиций различных категорий пациентов, так как в процессе консультирования возникает множество морально-этических проблем.
6.Медико-генетическое консультирование в профилактике наследственных заболеваний.
Медико-генетические консультации – один из видов специализированной медицинской помощи, суть которой состоит в диагностике наследственных заболеваний, в прогнозировании вероятности рождения больного ребёнка и помощи семье в принятии решения о деторождении.
Основные задачи медико-генетического консультирования включают:
1. установление точного диагноза наследственного заболевания;
2. определение типа наследования заболевания в данной семье;
3. расчёт риска повторения болезни в семье;
4. определение наиболее эффективного способа профилактики;
5. объяснение обратившимся смысла собранной и проанализированной информации, медико-генетического прогноза и методов профилактики.
В Костроме создана медико-генетическая консультация при центре репродукции и планировании семьи (ул. Свердлова).
Здесь ведут приём специалисты: врач-генетик, врач-гинеколог, врач-андролог, врач-психолог, есть там и лаборатория.
В медико-генетические консультации обращаются чаще всего молодые супруги, в родословной которых были случаи рождения детей с разными аномалиями. Врач-генетик на основе генеалогического метода попытается установить, является ли названное заболевание наследственным. Далее он определит тип наследования признака (если аномалия наследственна): аутосомно-доминантный, аутосомно-рецессивный, сцеплённый с полом; или характерный синдром при хромосомных изменениях в генотипе.
Затем врач рассчитает риск рождения ребёнка с аномалией. Степень риска рождения наследственно отягощённого ребёнка считается низкой от 0 до 5%, средней степени – до 12%, более 12% – высокой. При низкой степени риска врач рекомендует рождение ребёнка, при высокой – рекомендует воздержаться от деторождения.
При средней степени риска врач рекомендует женщине обратиться в медико-генетическую консультацию после наступления беременности для постановки диагноза плоду (метод пренатальной диагностики).
Методы ультрасонографии или ультразвукового скеннирования можно обнаружить у развивающегося плода нарушения анатомического строения органов и общих пропорций тела. Этим методом выявляют пороки развития опорно-двигательной системы. Раннее выявление таких аномалий как: мозговая грыжа, гидроцеоралия даёт возможность произвести аборт по медицинским показаниям и предотвратить рождение явно неполноценного ребёнка.
Внутриутробная диагностика возможна так же и с использованием метода амниоцентеза. С помощью шприца из матки производят забор небольшого количества аминотической жидкости вместе с живыми клетками плода, которые всегда в ней присутствуют. После культивирования этих клеток на искусственных питательных средах в них можно изучить кариотип и выявлять хромосомные и геномные мутации, определять пол плода, что важно для прогноза в отношении сцеплённого с полом наследования. Если обнаружится тяжёлая патология, врач рекомендует искусственное прерывание беременности.
 |  | ||
Грегор Мендель (1822-
Альбинос.
Хромосомы.
Хромосомы

Тест на дальтонизм
ЛИТЕРАТУРА
1. Ауэрбах Ш. Наследственность. – М., 1969.
2. Бадалян Л.О. Наследственные болезни у детей. - М., 1971.
3. Большая советская энциклопедия. Т. 7.- М., 1972.
4. Бочков Н.П. Генетика и медицина. – М., 1979.
5. Бочков Н.П. Генетика человека (Наследственность и
патология). – М., 1978
6. Дубинин Н.П. Генетика и человек. – М., 1978.
7. Инге-Вечтомов С.Г. Генетика с основами селекции. – М., 1989.
8. Карузина И.П. Учебное пособие по основам генетики. – М., 1980.
9. Киселёва З.С. Генетика. – М., 1983.
10. Козлова С.И. наследственные синдромы и медико-генетическое консультирование. – М., 1996.
11. Мюнтцинг А. Генетика. – М., 1967.
12. Полканов Ф.М. Мы и её величество ДНК. – М., 1968.
13. Фролов И.Т. Мендель, менделизм и диалектика. – М., 1972.
14. Шевцов И.А. Популярно о генетике. – Киев, 1989.
15. Encarta ’95. Компакт диск.
16. Encyclopaedia Britannica CD ’99 Компакт диск.
Похожие работы
... заболеваний Общие подходы к лечению наследственных болезней сходны с подходами к лечению болезней любой другой этиологии. При лечении наследственной патологии полностью сохраняется принцип индивидуализированного лечения (лечить не болезнь, а болезнь у конкретного человека). Этот принцип особенно важен, поскольку наследственные болезни обладают гетерогенностью и с одной и той же клинической ...
... Сфингомиелиназа Сфингомиелин, холестерин. Лейкоциты, материал биопсии органов, фибробласты кожи (инфантильная и ювенильная формы) Материал биопсии Гиперлипопротеидемии (ГЛП) Наследственные заболевания нарушений липидного обмена представлены большим количеством нозологических форм, которые обусловливают развитие атеросклероза и других форм сердечно-сосудистой патологии. Впервые ...
... . Эксперименты по «обратной мутации» проводятся только в микроорганизмах. Однако возможно, что в будущем генная инженерия будет исправлять ошибки природы и у человека. Пока основным способом борьбы с наследственными болезнями являются изменения условий окружающей среды, в результате чего развитие патологической наследственности становится менее вероятным, и профилактика путем медико-генетического ...
... число хромосом равно 48, а гаплоидное — 24. Усовершенствованные методы изучения хромосом позволили получить более точные сведения о количестве хромосом в клетках у человека, а также выявить аномалии нормального кариотипа, ответственные за некоторые уродства. Особенно плодотворным оказались два метода: 1. Обработка культуры клеток алкалоидом колхицином, который ведет к накоплению делящихся ...
0 комментариев