Навигация
Наследственные заболевания обмена веществ
70219
знаков
6
таблиц
1
изображение
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ
ГОМОЦИСТИНУРИЯ


К настоящему времени известно более 2500 наследственных ферментопатий, 20 классов наследственных болезней обмена, но только для части из них (для 300) установлен точный уровень метаболического блока и характер ферментативного дефекта. До сих пор большая часть этих заболеваний не диагностируется или выявляется поздно, когда происшедшие нарушения носят необратимый характер. Одна из трудностей ранней диагностики заключается в том, что в период новорожденности у этих детей нет специфических расстройств, а поздние проявления фенотипически схожи с заболеваниями ненаследственного генеза. Вторая особенность состоит в том, что для наследственных заболеваний обмена веществ характерен клинический полиморфизм, обусловленный генетической гетерогенностью. Это объясняется наличием множественных изоаллельных мутаций и возможностью возникновения мутаций в разных генах.
Клинические проявления наследственных болезней обмена веществ во многом определяются поражением нервной системы (особенно при нарушениях обмена аминокислот, липидов и кислых гликозамино-гликанов), что в свою очередь, усиливает имеющиеся нарушения и усугубляет тяжесть клинических проявлений заболевания. Для диагностики наследственных болезней важен анализ неврологических симптомов, особенно на ранних стадиях развития, и разграничение их от фенокопий- заболеваний ненаследственной природы со сходной клинической картиной. Важное практическое значение имеет выявление гетерозиготных носителей мутантного гена при наследственных болезнях. Например, при частоте фенилкетонурии 1:10 000, мутантный ген встречаетсяв популяции 1:50. Гетерозиготное состояние может клинически не обнаруживаться, поэтому выявление умеренно выраженных изменений метаболизма является единственным указанием на наличие мутантного гена.
Наследственные болезни обмена аминокислот
Роль аминокислот для организма человека чрезвычайно велика. Аминокислоты являются основными структурными элементами белков, необходимы для синтеза иммуноглобулинов, гормонов, служат источником энергии. Каждый фермент или белок имеет специфические свойства и функции, которые определяют и регулируют сложные обменные процессы и развитие организма.
Часть аминокислот не может синтезироваться в организме человека. Это незаменимые аминокислоты: триптофан, фенилаланин, метионин, лизин, лейцин, изолейцин, валин и треонин. В детском возрасте к их числу относится гистидин, т.к. организм ребенка не может синтезировать эту аминокислоту в необходимых для нормального роста количествах. Клетки растущих тканей содержат аминокислоты в высоких концентрациях, что является свидетельством высокой интенсивности процессов транспорта аминокислот через клеточные мембраны.
Для обеспечения нормального роста и развития важно не только количество поступающих аминокислот, но и их соотношение. При избытке или недостатке аминокислот развиваются явления аминокислотного дисбаланса. Например, избыток лейцина в пище тормозит рост организма, метионина- вызывает токсическое поражение нервной системы, цистина- способствует развитию жировой инфильтрации печени.
Таким образом, нарушения метаболизма аминокислот приводят к нарушению нормального функционирования организма человека.
Наследственные нарушения обмена аминокислот
(Ю.И. Барашнев, Ю.Е. Вельтищев, 1978 г.)
1. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их концентрации в крови и моче: фенилкетонурия, гистидинемия, триптофанурия, болезнь "кленового сиропа", орнитинемия, цитруллинемия и др. Наследование, в основном, по аутосомно-рецессивному типу. В основе развития заболеваний лежит нарушение синтеза или структуры тех или иных ферментов.
2. Наследственные нарушения обмена аминокислот, сопровождающиеся увеличением их выделения с мочой без изменения уровня в крови: гомоцистинурия, гипофосфатазия, аргиносукцинатацидурия и др. При данных энзимопатиях нарушено обратное всасывание в почках, что приводит к увеличению их содержания в моче.
3. Наследственные нарушения систем транспорта аминокислот: цистинурия, триптофанурия, болезнь Гартнепа и др. К этой группе относятся энзимопатии, развитие которых обусловлено снижением реабсорбции аминокислот в почках и кишечнике.
4. Вторичные гипераминоцидурии: синдром Фанкони, фруктоземия, галактоземия, болезнь Вильсона-Коновалова и др. При данных состояниях возникает вторичная генерализованная гипераминоацидурия в результате вторичных тубулярных нарушений.
Фенилкетонурия (ФКУ)
Впервые описана в 1934 г. Folling под названием "фенилпировиноградная имбецильность". Тип наследования - аутосомнорецессивный. Частота заболевания составляет 1:10000- 1:20000 новорожденных. Пренатальный диагноз возможен при использовании генетических зондов и биопсии ворсин хориона.
К развитию классической клинической картины при ФКУ приводит недостаточность фенилаланингидроксилазы и недостаточность редуктазы дигидроптерина- 2-го фермента, обеспечивающего гидроксилирование фенилаланина. Их недостаток приводит к накоплению фенилаланина (ФА) в жидких средах организма (схема 1). Как известно, ФА относится к незаменимым аминокислотам. Поступающий с продуктами питания и не используемый для синтеза белка, он распадается по тирозиновому пути. При ФКУ наблюдается ограничение превращения ФА в тирозин и, соответственно, ускорение его превращения в фенилпировиноградную кислоту и другие кетоновые кислоты.
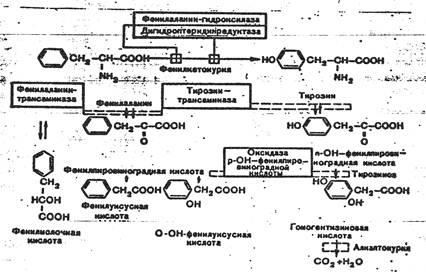
Схема 1. Варианты нарушений метаболизма фенилаланина.
Существование различных клинико-биохимических вариантов ФКУ объясняется тем, что фенилаланингидроксилаза является частью мультиферментной системы.
Различают следующие формы ФКУ:
1. Классическая
2. Скрытая.
3. Атипичная.
Развитие атипичных и скрытых форм ФКУ связывают с недостаточностью фенилаланинтрансаминазы, тирозинтрансаминазы и оксидазы парагидроксифенилпировиноградной кислоты. Атипичная ФКУ обычно не сопровождается поражением нервной системы в результате позднего развития ферментативного дефекта.
У женщин с фенилкетонурией возможно рождение детей с микроцефалией, задержкой умственного развития, нарушениями развития мочевыделительной системы, поэтому необходимо назначение диетотерапии во время беременности.
Клинические симптомы у больных ФКУ
При рождении ребенок с фенилкетонурией выглядит здоровым. Заболевание у этих детей проявляется на первом году жизни.
1. Интеллектуальный дефект. Нелеченный ребенок теряет около 50 баллов IQ к концу 1-го года жизни. У больных не выявляется зависимости между уровнем ФА и степенью интеллектуального дефекта.
2. Судорожный синдром (4 50%), экзема, гипопигментация.
3. Нарушение координации движения.
4. Задержка развития статических и двигательных функций.
5. Поражение пирамидных путей и стриопаллидарной системы. Клинические проявления классической ФКУ редко встречаются в странах, в которых действует программа неонатального скрининга на это заболевание.
У детей с фенилкетонурией наблюдается повышенный уровень в моче метаболитов ФА. Увеличение в физиологических жидкостях содержания ФА и недоокисленных продуктов его метаболизма приводит к поражению нервной системы. Определенная роль в этих нарушениях принадлежит дисбалансу аминокислот (дефицит тирозина, который в норме активно участвует в построении белкового компонента миелина). Демиелинизация является характерным патоморфологическим признаком фенилкетонурии. Нарушение соотношения аминокислот в
крови приводит к нарушению уровня свободных аминокислот в головном мозге, что вызывает слабоумие, гиперкинезы и другие неврологические симптомы.
Пирамидные симптомы обусловлены нарушением процессов миелинизации. Избирательный характер поражения нервной системы объясняется особенностями миелинизации- поражаются наиболее молодые в филогенетическом отношении отделы, выполняющие сложные и дифференцированные функции. С недостаточным образованием меланина из тирозина связывают голубой цвет глаз, светлую кожу. Запах "плесени" ("мышиный", "волчий") объясняется наличием фенилуксусной кислоты в моче. Кожные проявления (экссудативный диатез, экземы) связаны с выделением аномальных метаболитов. Недостаточность образования адренергических гормонов из тирозина приводит к артериальной гипотонии.
Необходимо отметить, что при ФКУ в патологический процесс вовлекается печень, но характер морфологических расстройств не является специфичным: выявляются признаки тканевой гипоксии, нарушения окислительной и белоксинтезирующей функции, перегрузка липидами. Наряду с этим наблюдаются компенсаторно-приспособительные изменения: высокое содержание гликогена, гиперплазия митохондрий. Генерализованную гипераминоацидемию при ФКУ можно объяснить вторичным нарушением метаболизма аминокислот в связи с повреждением гепатоцитов, т.к. многие ферменты, участвующие в аминокислотном обмене, локализуются в печени.
У нелеченых больных с классической ФКУ наблюдается значительное снижение концентрации катехоламинов, серотонина и их производных в моче, крови, ликворе. Поэтому в комплексном лечении ФКУ необходима промедиаторная коррекция, так как парциальный интеллектуальный дефект может быть связан с нейромедиаторными нарушениями.
Критерии диагностики классической формы фенилкетонурии:
1. Уровень ФА в плазме выше 240 ммоль/л.
2. Вторичный дефицит тирозина.
3. Повышенный уровень в моче метаболитов ФА.
4. Сниженная толерантность к полученному внутрь ФА.
Методы диагностики фенилкетонурии:
1. Проба Феллинга с FeCl3- при положительном анализе появляется сине-зеленое окрашивание мочи.
2. В крови выявление избытка фенилаланина возможно с помощью бактериального экспресс- теста Гольдфарба или теста Гатри (т.к. в течение первых дней жизни фенилпировиноградная кислота в моче может отсутствовать).
При ФКУ проводится лечение диетой с ограниченным содержанием ФА (главным образом назначают овощные блюда, мед, фрукты). Такие продукты, как молоко, молочные изделия, яйца, рыба, должны быть полностью исключены в период пребывания больных с ФКУ на острой диете. Назначаются специальные препараты (цимогран, лофеналак) и витамины.
Оптимальные сроки обследования новорожденных- 6-14 день жизни, начало терапии - не позднее 21 дня жизни. Необходимо помнить, что проведение исследования в первые сутки не исключает ложноположительных или ложноотрицательных результатов ( повторное исследование проводят до 21 дня жизни ). Эффективность лечения оценивается по интеллектуальному уровню развития пациента. Необходимо отметить, что лечение, начатое после года не нормализует интеллект полностью (возможно, это связано с развитием необратимых изменений в мозге).
ГОМОЦИСТИНУРИЯ
Заболевание впервые описано в 1962 г. Carson и Neil. Наследуется по аутосомно-рецессивному типу. Частота гомоцистинурии составляет 1:200 000 новорожденных.
В основе заболевания лежит отсутствие или снижение активности фермента цистатионинсинтетазы, что ведет к нарушению обмена метионина. Кофактором цистатионсинтетазы является витамин В6. Поэтому наблюдается пиридоксинчувствительная и пиридоксинрезистентная формы. У родителей и родственников больных часто обнаруживают шизофрению. Рядом авторов отмечается фенотипическое сходство с болезнью Марфана. Однако, при гомоцистинурии в отличие от болезни Марфана более выражены изменения нервной системы, снижение интеллекта и судорожный синдром.
Формы гомоцистинурии:
Похожие работы
... ─ и конкордантные, и дискордантные (как и в семье из двух детей). Наследственные болезни подразделяются на хромосомные и генные болезни (наследственные заболевания обмена веществ, наследственные нарушения иммунитета, болезни с преимущественным поражением эндокринной системы и др.) Хромосомные болезни. Характеризуются изменением структуры или числа хромосом. Они встречаются примерно у 1-2 ...
... телец (лейкоцитов). Отмечено значение генных мутаций и при возникновении лейкозов (белокровии), что доказывается путем анализа анамнеза семей, в которых выявлены больные. Кроме того, доказательством роли наследственного фактора в возникновении лейкозов является частота заболевания у близнецов, а также большая частота врожденного лейкоза при хромосомных заболеваниях, в частности болезни Дауна. К ...
... , что существуют сходные врождённые болезни обмена веществ со своим специфическим боихимическим дефектом. В настоящее время в биохимической генетике описано более 300 - 19 - наследственных болезней обмена веществ с изученой аномалией. В клинической практике для биохимической диагностики известных болезней обмена веществ применяют систему качественных и ...
... их зрение снижено и не улучшается с возрастом. Альбинизм встречается с частотой 1 на 39.000, наследуется по аутосомно-рецессивному типу (см. приложение рис. 1). Ген локализован на длинном плече 11-й хромосомы. Наследственные заболевания, связанные с нарушением обмена углеводов Известно, что углеводы входят в состав ряда биологически-активных веществ — гормонов, ферментов, мукополисахаридов ...
0 комментариев