Цели и задачи изучения темы «Молекулярные перегруппировки»
Применение электронно-дидактических средств в обучении химии
Общие представление о перегруппировки. Перегруппировка Бекмана является нуклеофильной, внутримолекулярной, стереоспецефичной
Механизм перегруппировки
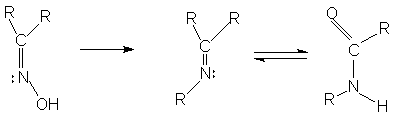
При отщеплении от диазометилкетона (1) азота под влиянием катализатора или температуры образуется промежуточный бирадикал(2)- карбен
Общие сведения о перегруппировке
Доказательства электрофильности перегруппировки
Общие сведения о перегруппировке
Механизм перегруппировки гидроксил амина
Использование тестов для оценки знаний
Навигация
Общие представление о перегруппировки. Перегруппировка Бекмана является нуклеофильной, внутримолекулярной, стереоспецефичной
Компьютерные технологии при изучении темы "Молекулярные перегруппировки"
114347
знаков
2
таблицы
66
изображений
1. Общие представление о перегруппировки. Перегруппировка Бекмана является нуклеофильной, внутримолекулярной, стереоспецефичной.
2. Механизм перегруппировки.
Превращение кетоксимов в амиды кислот, протекает по SN1 механизму, в присутствии PCI5; POCI3; H2SO4; HCI; т.е. кислоты Льюиса и все протонные кислоты.
К атому азота мигрирует группа R, находящаяся в анти- положении к группе –ОН, т.е.
т.е. происходит разрыв N-O связи с одновременной миграцией анти- расположенной к группе –OH, т.е.

например: 2-хлор-5-нитробензофеноксимов при действии щелочи легко отщепляет HCI, образуя изоксазольный цикл
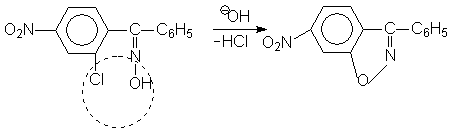
т.е. в этом изомере –ОН группа и CI замещенное бензойное кольцо находится в цис- положении. Механизм перегруппировки следующий:
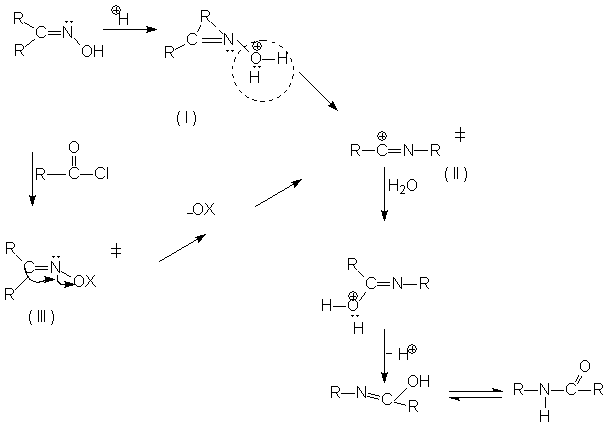
В присутствии сильных кислот перегруппировка протекает путем протонирования оксима с образованием соединения (I), с последующим отщеплением воды и образования катиона (II), при действии хлорангидридов образуются промежуточные эфиры (Ш) в которых ОХ хорошо уходящая группа, образуется промежуточное соединение (II).
Ш превращается в II без кислот в нейтральных растворителях, чем сильнее кислота ХОН, тем быстрее происходит перегруппировка. Отщепление НОН и миграция R одновременно (интрамолекулярно).
Эта реакция первого порядка, ускоряется полярными растворителями
и облегчается при возрастании кислотности реакционной среды, облучение на него не действует.
4. Доказательство стереоспецифичности
Система C=N является жесткой и подвижность её ограничена, то атака будет происходить легче со стороны, в которой нет заместителя.
Атака радикала R будет затруднена стерическими факторами, т.е. у атома азота находится Н-О-Х (хорошо уходящая группа):

Атака будет происходить со стороны заместителя R у атома N2, т.к. не имеется других заместителей. На основании этого можем сделать вывод, что реакция является стереоспецифичной.
Бензильная перегруппировка![]()
Либих (1838 – 1839г)
Общие сведения о перегруппировке
Бензильная перегруппировка основана на превращении α- дикетонов под действием щелочей в α- оксикислоты[14]
Бензильная перегруппировка является нуклеофильной, интрамолекулярной, не стереоспецифической, протекает без изомеризации в алифатическом ряду с миграцией от одного атома углерода к другому атому углерода.
Механизм перегруппировки.
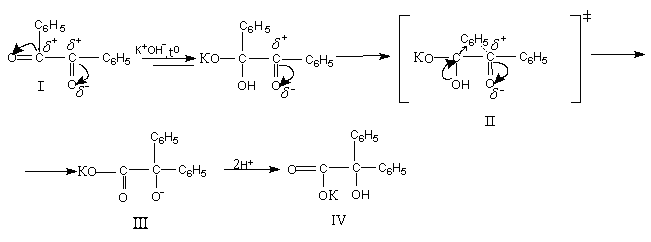
Впервые она была описана Ю.Либихом в 1838 году на примере бензила, который при нагревании со спиртовой щелочью превращается в бензиловую кислоту:
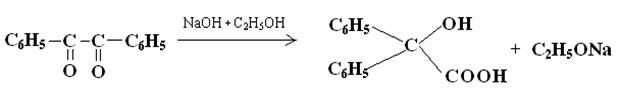
1.Первая стадия перегруппировки – это процесс превращения бензила (I) под действием щелочи в калиевую соль бензиловой кислоты (II). Ввиду большей электроотрицательности атома кислорода на нем сосредотачивается вся электронная плотность, за счет чего на атоме углерода возникает частичный положительный заряд. Если данное соединение провзаимодействует с щелочью, то образуется неустойчивое соединение – калиевая соль бензиловой кислоты. Данный процесс (переход от I к II) можно охарактеризовать как нуклеофильное бимолекулярное присоединение, т.к. эта стадия является лимитирующей, то основание, являющееся нукклеофилом, атакует электрофильный центр с разрывом двойной связи.
Таким образом происходит присоединение по карбонильной группе. А скорость будет описываться кинетическим уравнением 2 порядка:
V = к ·[бензил]· [ОН-]
2.Вторая стадия данной перегруппировки - это гетеролитический отрыв фенильного радикала с дальнейшим присоединением его к соседнему атому углерода, несущему дефицит электронной плотности. В результате того, что разрыв и образование новой связи происходят синхронно (одновременно), этот процесс является внутримолекулярным (интрамолекулярным).
3.Третьей стадией является стадия протонизации, за счет чего открытый анион стабилизируется и образуется бензиловая кислота (IV), т.е. происходит специфический катализ.
Таким образом, по предмету данная перегруппировка называется: превращение бензила под действием щелочи в бензиловую кислоту.
РЕТРОПИНАКОЛИНОВАЯ ПЕРЕГРУППИРОВКА
1.Общие сведения о перегруппировке.
Ретропинаколиновая перегруппировка основана на образовании двух третичных атомов углерода из одного четвертичного. Данная перегруппировка является нуклеофильной, интрамолекулярной, стереоспецифической, протекает с изомеризацией в алифатическом ряду с миграцией от атома углерода к другому атому углерода.В 1901 г. Н. Д. Зелинский при дегидратации кислотой пинаколинового спирта вместо ожидаемого 2,2-диметилбутена-З получил 2,3-диметилбутен-2:
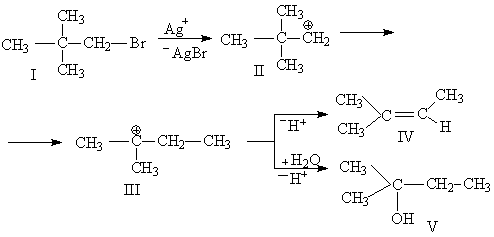

Поскольку здесь, в противоположность пинаколиновой перегруппировке, скелет из пинаколинового превращается в симметричный, как у пинакона, перегруппировки этого типа называются ретропинаколиновыми или иногда перегруппировками Вагнера - Меервейна, так как первой разъясненной перегруппировкой этого типа была открытая Е. Е. Вагнером в 1899 г. перегруппировка пинена в хлористый борнил, впоследствии (1910 г.) подробно изученная Меервейном и сопоставленная им с простейшими примерами этого типа перегруппировок.(15)
Доказательства стереоспецифичности перегруппировки.
На гетеролитический и, более того, ионный характер этих перегруппировок указывает то, что они протекают гораздо быстрее в сольватирующих растворителях с высокой диэлектрической проницаемостью. Как и для разных типов пинаколиновых перегруппировок, покидающая молекулу группа может быть не только гидроксилом, но и галогеном, азотом алифатического диазония и сложноэфирной группой, например тозилатной..
Нельзя считать доказанным, что во всех ретропинаколиновых перегруппировках не фигурирует свободный карбкатион: стереохимическое течение реакции известно не везде. Напротив, во многих случаях вполне вероятны равновесия, подобные XI«XII. Некоторым аргументом в пользу участия свободного катиона может служить то, что в большинстве случаев результаты перегруппировки указывают на предпочтительное направление реакции в сторону образования более устойчивого карбкатиона, т. е. с более компенсированным положительным зарядом. Так, из катионов II и III более устойчив последний, так как его положительный заряд подавляется +I-эффектом трех алкильных групп, тогда как в катионе II ,+I-эффектом всего одной трет-бутильной группы. Вообще по этой причине карбкатионы с зарядом на третичном углероде устойчивее катионов с зарядом на вторичном, а последние устойчивее карбкатионов типа -СН2+. Однако в случае R = C6H5 формулы XI и XII предложено заменить на XV - промежуточное соединение - фенониевый катион с равновероятным раскрытием цикла при действии на него воды и образованием соединений XIII и XIV.
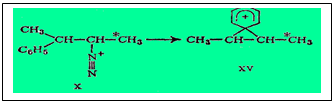
Понятие о фенониевом катионе впервые ввел Крам с целью объяснить стереоспецифическое поведение в ретропинаколиновои перегруппировке двух диастереоизомеров З-фенил-2-бутилтозилата.
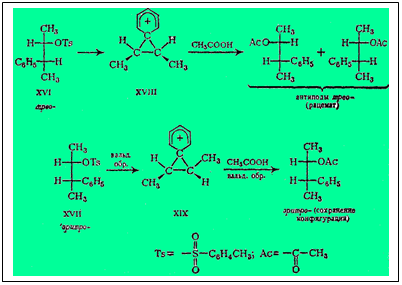
При ацетолизе трео-стереоизомер XVI полностью рацемизуется, а эритро-стереоизомер XVII превращается в ацетат, сохраняющий в полной мере оптическую активность. Крам постулирует образование в качестве промежуточных форм в течение перегруппировки структур XVIII и XIX, включающих фенониевый катион, привязанный симметрично к обоим углеродным атомам - стартовому и финишному.
Углеродный атом ароматического кольца, связанный первоначально со стартовым атомом С, в трехчленной промежуточной форме стал тетраэдрическим. Значит, плоскость бензольного кольца стала перпендикулярной к плоскости чертежа. Положительный заряд финишного атома С теперь рассредоточен в триаде углеродных атомов трехчлена, а следовательно, и по всем атомам бензольного цикла, что, по Краму, снижает энергетический уровень этой переходной формы и тем самым ускоряет реакцию.
Разрыв под действием уксусной кислоты гипотетического трехчленного катиона в случае трео-изомера XVI, имеющего плоскость симметрии, должен вести к рацемату, так как разрыв равновероятен слева и справа (или, иначе, две симметричные молекулы при реакции не могут дать одну оптически активную форму, но лишь рацемат). Напротив, в случае эритро-изомера XVII ацетолиз несимметричной трехчленной структуры, с какой бы стороны ни шла атака уксусной кислоты, приводит к исходной конфигурации.
Следует обратить внимание на то, что и замыкание в трехчленный Никл с удалением тозилат-аниона, и разрыв трехчленного цикла с возвращением фенониевого катиона в бензоидную форму происходит с вальденовским обращением. В. Хюккель, оспаривающий трактовку Крама и самое существование фенониевого катиона, и трехчленного цикла, указывает, что стереоспецифичность рассматриваемой реакции можно объяснить просто переменой местами фенильной группы с тозилатной (или с заменяющей ее ацетатной) с двумя вальденовскими обращениями:
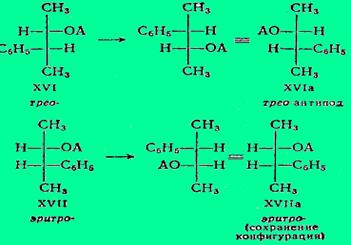
Здесь ОА обозначает как уходящий тозилат-анион, так и вступающий (в опытах Крама) ацетат-анион.
Само собой разумеется, что в условиях реакции ацетат XVIa может перегруппировываться снова в ацетат XVI, так что рацемизацию обусловливают оба процесса XVI«XVIa.
Возражения Хюккеля основательны, и широко распространенная, ныне концепция фенониевого иона как промежуточной формы в перегруппировках не может считаться, бесспорно принятой в науке.
Реакции, где мигрирует ароматическая группа, а не алкил или водород, по мнению сторонников гипотезы фенониевого катиона, протекают в сотни и тысячи раз быстрее именно благодаря относительно низкому энергетическому уровню фенониевого катиона с его рассредоточенным по многим атомам положительным карбониевым зарядом. Возражение Хюккеля в этом случае состоит в том, что структуры типа XVIII и XIX по существу являются «классическими» и никак не могут рассматриваться как переходное состояние, а скорость реакции определяется только энергетическим уровнем переходного состояния. В самом деле, стабильные катионы подобного типа мы встречали, например, в виде

при описании реакции Фриделя - Крафтса. Таким образом, структуры XVIII и XIX отделены от начальных и конечных продуктов реакции энергетическими барьерами, которые и определяют константу скорости превращений исходного XVI в промежуточный продукт и последнего в продукт XVIa и соответственно XVII в промежуточный продукт и далее - в XVIIa. Конечно, можно выдвинуть контрвозражение, что если оба переходных состояния, отделяющие промежуточный продукт, близки к нему по геометрии и структуре, то они и энергетически близки. Тогда энергетические барьеры, отделяющие промежуточный продукт, невысоки, а следовательно, энергетический уровень этого продукта все же приблизительно определяет скорость реакции (постулат Хэммонда).
В 1967 г. Олах описал эксперимент, который, казалось бы, непосредственно доказывает существование фенониевых ионов по крайней мере в некоторых случаях. Оказалось, что п-(2'-хлорэтил) -анизол, растворенный в избытке SbF5 при - 80 °С, дает острый одиночный пик (синглет) в спектре ЯМР в области метиленовых протонов. Это означает, что обе метиленовые группы становятся эквивалентными после удаления хлор-аниона, т. е. после образования карбониевого иона:
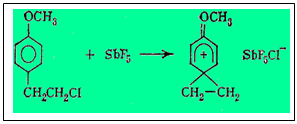
Если бы метиленовые группы были неэквивалентны, то в спектре ЯМР в этой области было бы два триплета, относящихся к разным метиленовым группам. Однако на деле эти данные не исключают быстрой обратимой изомеризации «неклассического» карбкатиона, т. е. равновесия типа:
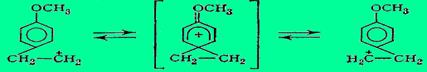
При этом скорость взаимопревращений классических карбкатионов должна быть настолько велика, чтобы ЯМР не мог уловить каждый карбкатион как отдельную частицу.
Сопоставляя теоретические соображения Хюккеля и экспериментальные данные Брауна, можно сказать, что «неклассический» катион правильнее представлять не как самостоятельно существующую частицу и не как сумму резонирующих «классических» катионов, а как переходное состояние перегруппировки, которое находится не в минимуме, а в максимуме потенциальной кривой (на вершине барьера, разделяющего два переходящих друг в друга «классических» катиона I и II).
Альдегидо - кетонная перегруппировка
1.Общие сведения о перегруппировке
Альдегидо- кетонная перегруппировка основана на превращении альдегидов в изомерные кетоны, сопровождающаяся обменом водорода альдегидной группы на соседний углеводородный радикал под действием протонных кислот (кислот Льюиса, серной и т. д. )
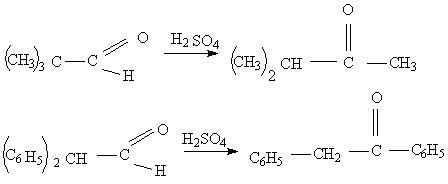
Альдегидо- кетонная перегруппировка является нуклеофильной, интрамолекулярной, стереоспецифической, протекает с миграцией от атома углерода к другому атому улерода (от С-С) , с изомеризацией в насыщенных системах.(16)
2.Механизм перегруппировки.
Механизм превращения альдегидов в кетоны сходен с механизмом ретропинаколиновой перегруппировки и может быть представлен следующей схемой:
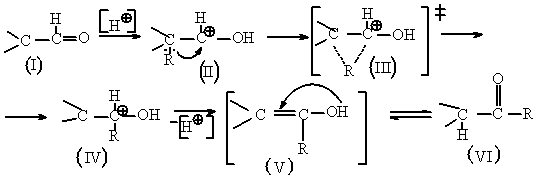
1.Благодаря неподеленной паре электронов на атоме кислорода альдегиды обладают основными свойствами и реагируют с кислотами.
При взаимодействии с протонными кислотами альдегиды (I) присоединяют протон, вследствии чего образуется σ – связь ОН(II.) В результате возрастает электрофильность углеродного атома карбонильной группы(17).
2.Т.к. α- углеродный атом в соединении (II) связан с радикалом, который за счет резонансных факторов, эффекта гиперконьюгации, имеет неподеленную пару, то данная пара начинает выступать в качестве нуклеофила. При этом начинает образовываться связь между радикалом, находящимся у α- углеродного атома и карбкатионным углеродом, при этом одновременно происходит разрыхление связи между α- углеродным атомом и радикалом при нем. В результате образуется неустойчивое соединение (III).
3.В результате происходит образование вторичного катиона на α- углеродном атоме (IV).В дальнейшем происходит регенирация протона с образованием соединения (V),которое по правилу Эльтекова- Эйленмеера является неустойчивым, т.к. образуется система, в которой у одного атома углерода находятся двойная связь и гетероатом, несущий неспаренную пару электронов.
4.Таким образом, соединение (V), вследствии своей неустойчивости изомеризуется с образованием кетона.
Пинаколиновая перегруппировка
1.Общие сведения о пергруппировке.
Катализируемая минеральными кислотами перегруппировка 1, 2- диолов называется пинаколиновой перегруппировкой (18). Данная перегруппировка является нуклеофильной, интрамолекулярной, протекает без изомеризации в алифатическом ряду с миграцией от атома углерода к другому атому углерода. Движущей силой реакции является отщепление молекулярного азота, а содействующим фактором - легкий отрыв протона от кислорода, что в итоге приводит к стабильной молекуле.
2.Механизм перегруппировки.

Многочисленные исследования различных замещенных гликолей и 1,2- аминоспиртов показали, что мигрирующая группа (Н, алкил, арил, галоген) выполняет роль нуклеофильного реагента, атакуя (+) заряженный атом углерода, возникающий после отщепления одного из протонированных гидроксилов.(19)
При этом установлено, что перескакивающий заместитель подходит к атому углерода всегда со стороны, противоположной положению потерянного гидроксила. Это означает, что в карбкатионе вращение С-С- связи отсутствует, вероятно, вследствии сильного донорно- акцепторного взаимодействия оставшегося –ОН с носителем (+)- заряда. Мигрирующая группа (Н, СН3, С6Н5 и др.) в переходном состоянии связана с обоими атомами углерода трехцентровой связью. Второй гидроксид- ион, сыграв свою активную роль стабилизатора карбкатиона вновь возвращается к своему (С1) атому углерода, но уже в виде протонированой С=О- группы.
Перегруппировка требует энергетических затрат на разрыв одной сигма- связи (С1- R) и перераспределение электроной плотности между химическими связями. Установлено, что результат перегруппировки для несимметрично замещенных гликолей сильно зависит от конформации заместителей и всей молекулы в целом.
На основании выше сказанного можно сделать вывод, что рассмотренная перегруппировка является нуклеофильной, стереоспецифической, внутримолекулярной.
Расчленение на стадии здесь дано искусственно, для ясности.
Подробно изучено, какие радикалы в этой перегруппировке легче мигрируют. В порядке легкости миграции радикалы располагаются в следующий ряд:
п-CH3OC6H4 > п-CH3C6H4 > п-ClC6H4 > Алкилы > H
Общий вывод таков: легче мигрирует радикал, в наибольшей степени рассредоточивающий положительный заряд, появившийся в системе в результате отрыва -ОН, или, другими словами, легче мигрирует радикал, наиболее способный подвергнуться электрофильной атаке. То обстоятельство, что начальным моментом перегруппировки является отрыв гидроксильного аниона и образование карбониевого катиона (в переходном состоянии тотчас рассредоточивающего свой заряд между С, от которого оторван гидроксил, соседним С скелета и ключевым С мигрирующей R'-группы), доказывается следующими аналогиями:

(Здесь даны только исходный бромгидрин и конечный пинаколин; механизм, очевидно, тот же.)
Еще одна аналогичная перегруппировка - «окислительное дезаминирование» Мак-Кензи:
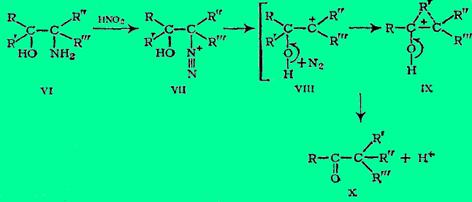
Движущей силой реакции является отщепление молекулярного азота, а содействующим фактором - легкий отрыв протона от кислорода, что в итоге приводит к стабильной молекуле.
Общий механизм реакции можно выразить одной схемой синхронного превращения:

3.Доказательства стереохимического течения перегруппировки.
На примере окислительного дезаминирования впервые установлено и стереохимическое течение перегруппировки пинаколинового типа. Мигрирующий радикал R', конечно, если с углеродом, несущим гидроксил, связан асимметрический атом этого радикала, сохраняет свою (правую или левую) конфигурацию. Группировка же вокруг углеродного атома, к которому мигрирует R', претерпевает вальденовское обращение (что можно констатировать, конечно, только если исходный аминоспирт был выделен в стереохимически индивидуальной форме). Все это безусловно доказывает, что промежуточный карбкатион не появляется в свободном состоянии: карбкатионы имеют плоскую конфигурацию, и когда они фигурируют как промежуточные стадии реакции, т. е. когда реакции разыгрываются по механизму SN1, обычно происходит полная или далеко идущая рацемизация.
Таким образом, стереохимическое течение пинаколиновых перегруппировок такое же, как в реакциях SN2. Это как бы SN2.-замещение групп ОН или Вг (в окислительном дезаминировании - диазониевой группы N2+) у несущего эти группы углеродного атома на радикал R' от соседнего углерода.
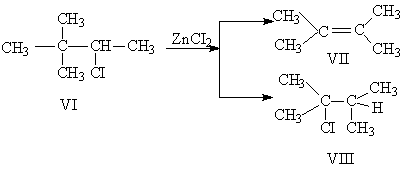
В первых двух случаях выведению групп ОН и Вг благоприятствует действие Н+ или ZnCl2 (льюисова кислота) и Ag+, в третьем случае перегруппировке содействует самопроизвольный распад непрочного алифатического диазония. Пинаколиновой перегруппировке содействует также +T-эффект гидроксила, благодаря которому реализуется превращение спиртовой группы пинакона в кетонную группу пинаколина. Следует еще пояснить формулы IV и IX. Они изображают переходное состояние, в котором R' еще не оторвался от материнского углерода, но уже начал связываться с углеродом, принимающим мигрирующую группу. Оба эти углерода и R' связаны трехцентровыми электронными орбиталями. Если R' = Н, то речь идет о гидридном перемещении. Следует, однако, напомнить, что, по данным Тиффено, Орехова и Бахманна, водород мигрирует труднее, чем алкилы или арилы.
В отношении радикала R' процесс должен рассматриваться как электрофильное замещение связанной с ним группы:

4.Применение перегруппировки.
Пинаколиновые перегруппировки многократно использовались для изменения структуры алициклов, расширения или сужения цикла. Например:

Н. Д. Зелинский и Н. И. Шуйкин осуществили следующий синтез спиранового кетона:

Направление реакций в сторону образования производного циклогексанона или циклопентильного кетона зависит, в частности, от характера R, но также и от стереохимии цикла.
Поскольку перегруппировка происходит с вальденовским обращением у принимающего углерода, мигрирующий радикал должен поступать со стороны, противоположной уходящей группе (НО, Вг, N2+ в приведенных выше примерах). В случае фиксированного в цикле пространственного расположения этих групп по отношению к остающемуся гидроксилу, углеродному радикалу и СН2-группе цикла геометрический фактор сказывается и иногда направляет реакцию по-разному. Пример: {picture8} Пинаколиновая перегруппировка проходит только для цис-аминоспирта (диазотированного азотистой кислотой): транс-аминоспирт сохраняет свой скелет
Перегруппировка Демьянова
1.Общие сведения о перегруппировке.
Перегруппировка Демьянова относится к перегруппировке в ненасыщенных системах, основанной на расширении или сужении цикла на один атом углерода при превращении алициклических или гетероциклических первичных аминов в спирты под действием азотистой кислоты (20). Данная перегруппировка является внутримолекулярной ( интра- ), т.е. разрыв старых связей и образование новых происходит синхронно; является нуклеофильной; протекает с миграцией от атома углерода к другому атому углерода (от С-С); протекает с участием карбкатиона.
При рассмотрении перегруппировки Демьянова следует учитывать устойчивость циклов, то есть то, что шестичленные и пятичленные циклы энергетически наиболее устойчивы чем трехчленные, четырехчленные и т.д. Поэтому при рассмотрении расширения или сужения циклов в результате перегруппировки следует иметь в виду теорию устойчивости циклов.
Так, например, при действии азотистой кислоты на циклогексилметиламин (I) образуется смесь циклогексилкарбинола I(III) и циклогептанола (смесь изоборнеона) (IV).
Эта же смесь спиртов образуется и при действии азотистой кислоты на циклогептиламин (II):

2.Механизм перегруппировки.
Механизм перегруппировки Демьянова аналогичен механизму пинаколиновой и ретропинаколиновой перегруппировок.
Так, образование циклогексилкарбинола при действии азотистой кислоты на циклогексилметиламин (I) может быть объяснено следующей схемой:

| |
Однако карбониевый ион (V) может претерпевать перегруппировку типа ретропинаколиновой, в результате чего возникает карбониевый катион (VI), а из него- циклогептанол:

|
Т.е. пара электронов как нуклеофил атакует атом углерода, на котором находится недостаток электронной плотности, причем образование новой связи и разрыв старой происходит синхронно, т. е. перегруппировка является внутримолекулярной.
При действии азотистой кислоты на циклогептиламин образуется карбониевый катион (VI), который реагирует с водой, дает циклогептанол:
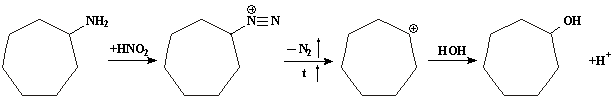
А претерпевая перегруппировку типа пинаколиновой, образует карбониевый ион (V), и из него циклогексилкарбинол:
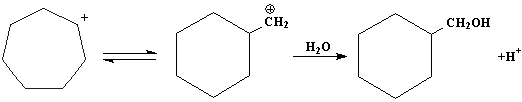
3.Побочные реакции.
Перегруппировка Демьянова может сопровождаться образованием олефинов за счет отщепления протона от карбониевых ионов (V), (VI) или (VII):
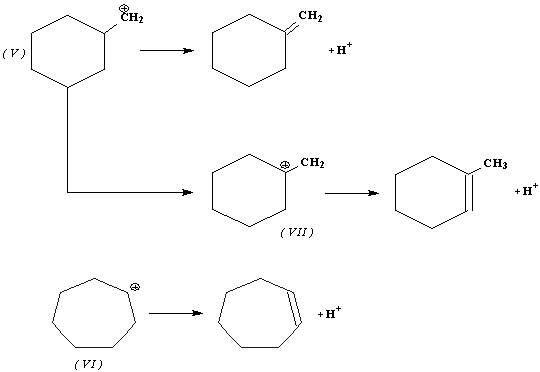
Реагирует ли азотистая кислота с циклогексилметиламином или циклогептиламином, в реакционной смеси устанавливается равновесие между карбониевыми ионами 5 и 6. Это является причиной образования одинаковой смеси спиртов в обоих случаях. Таким образом, в данной перегруппировке побочная реакция-образование циклических олефинов.
Сужение и расширение циклов наблюдается не только при реакции олефинов с азотистой кислотой, но и в других случаях, когда образуются карбониевые ионы.
Н.Я.Демьянов и Н.М .Кижнер установили, что при действии бромистоводородной кислоты на циклопропилкарбинол образуется циклопропилбромметан и бромциклобутан, из циклобутанола при действии бромистоводородной кислоты получается циклопропилкарбинол:
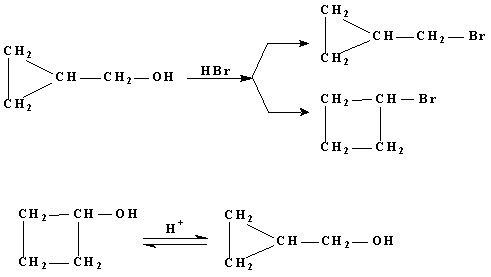
Перегруппировка Наметкина.
1.Общие сведения о перегруппировке.
Переход α-метилкамфена в 4-метилизоборнеол в условиях кислотного катализа называется перегруппировкой Наметкина, иначе ее называют камфеновой перегруппировкой второго рода. Данная перегруппировка наблюдается в ряду терпенов.(21)
2.Механизм перегруппировки.
Камфеновая перегруппировка второго рода была открыта Наметкиным и Брюсовой.

Механизм перегруппировки Наметкина заключается в изомеризации углеродного скелета по типу ретропинаколиновой или, при обратном процессе, - пинаколиновой перегруппировки.
Рассмотрим механизм:
1.К α-метилкамфену (I) идет присоединение воды по двойной связи в соответствии с правилом Марковникова (II).
2.Дальнейшая протонизация дает оксониевый ион (III). Как известно, вода – это хорошо уходящая группа. Вследствие этого, соединение (III) превращается в открытый карбкатион (IV).
3.Затем собственно здесь начинается перегруппировка: атака нуклеофила (пары электронов) по положительному реакционному центру. То есть миграция группы, имеющей избыток электронов (÷СН3) к атому, несущему положительный заряд (карбкатион), стабилизация карбкатиона, одновременный уход ÷СН3 группы и атака его по карбкатиону обуславливает тот факт, что данная перегруппировка является внутримолекулярной (интрамолекулярной).
4. В месте ухода÷СН3 группы появляется новый реакционный центр, который может сразу перейти в конечный продукт (4-метилиззоборнеол), либо сразу при взаимодействии с водой, либо претерпевающий перегруппировку (VII).
Таким образом, перегруппировка Наметкина является нуклеофильной, внутримолекулярной перегруппировкой.
3.Зависимость продуктов перегруппировки от различных факторов.
Состав и структура продуктов камфеновой перегруппировки определяется относительной скоростью ряда параллельных изомеризации. Например, для производных борнеола (VII) возможны: 1,2-гидридный сдвиг (а), 142-миграция группы СН3 (б), перемещение мостика (в) и сужение алицикла (г). Однако в основном реализуется путь (г), поскольку в случае (а) образуется менее стабильный вторичный карбкатион (IX), путь (б) запрещен Бредта правилом, по пути (в) получается термодинамически невыгодный четырехчленный цикл:
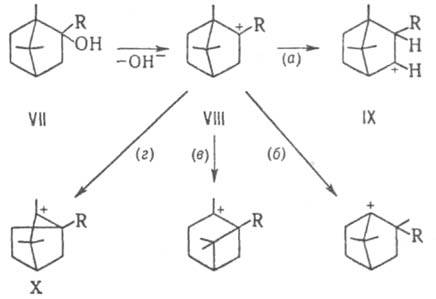
Карбкатион X может далее элиминировать протон, присоединить нуклеофил или претерпеть перегруппировку, например 1,2-сдвиг мостиковой группы СН3. Камфеновую перегруппировку применяют для синтеза различных производных терпенового ряда, например, камфоры из о-пинена, изоборнилацетата из камфена. Камфеновая перегруппировка I рода открыта Е. Е. Вагнером в 1899, II рода - С. С. Наметкиным в 1924.
Перегруппировка Вагнера –Меервейна
1.Общие сведения о перегруппировке.
Перегруппировка Вагнера –Меервейна является нуклеофильной, интрамолекулярной, стереоспецифической, протекает в ненасыщенной системе без изомеризации с миграцией от атома углерода к другому атому углерода. (22)
Перегруппировки Вагнера-Меервейна, общее название реакций, протекающих с 1,2-миграцией группы R(H, алкил или алициклич. фрагмент, арил, ацил, алкоксикарбонил и др.) к карбкатионному центру, возникающему в молекуле при нуклеофильном замещении, присоединении к кратной связи или элиминировании:
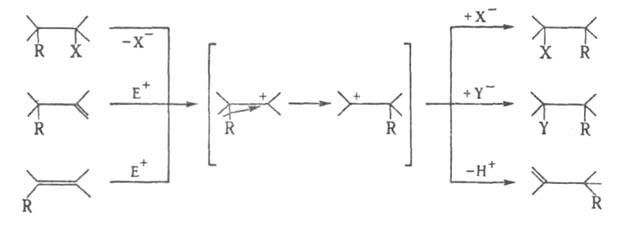
где Х-уходящая группа, например ОН, Nj, Hal, Y~ -внешний нуклеофил, Е+ - электрофильный агент, например Н + , AlkCO + .
Перегруппировку Вагнера-Меервейна относят к секстетным перегруппировкам и подразделяют на пинаколиповые и ретропинаколиновые перегруппировки. Родственной этой перегруппировке является Демьянова перегруппировка.
Перегруппировку Вагнера-Меервейна претерпевают, как правило, углеводороды и их производные, имеющие разветвленный углеродный скелет. В ряду алициклических соединений часто сопровождается расширением или сужением цикла. С данной перегруппировкой конкурируют другие реакции карбкатионов-фрагментация, депротонирование, присоединение нуклеофила. Протеканию перегруппировки способствуют увеличение диэлектрической проницаемости растрителя и уменьшение его основности, а также прочное связывание уходящего аниона, напр. в виде комплексных анионов А1С14-, SbClg.
Похожие работы
... которых можно осуществить превращения:→ 3.1. этанол → бутадиен-1,3 → бутадиеновый каучук 3.2. 2-метилбутан → изопрен → цис-полиизопрен ГЛАВА IV. ТЕСТЫ И ЗАДАЧИ ПО ТЕМЕ «НЕПРЕДЕЛЬНЫЕ УГЛЕВОДОРОДЫ» Задачи 1. При пропускании 11,2 л (н.у.) смеси этана, этилена и ацетилена через склянку с бромной водой масса склянки увеличилась на 10,9 г. При пропускании исходной ...

... в химию магнитные взаимодействия. Будучи пренебрежимо малыми по энергии, магнитные взаимодействия контролируют химическую реакционную способность и пишут новый, магнитный «сценарий» реакции. Дизайн молекулярных магнетиков — одно из новых научных направлений современной химии, связанное с синтезом систем высокой размерности. Сегодня достижения современной химии таковы, что химики могут ставить ...
... новые возможности, которые возникали с появлением новых методов исследования, позволяли делать открытия, радикально менявшие взгляды на патологию, начинать качественно новые этапы её развития. Патологическая анатомия использует три основных метода исследования — вскрытие трупов людей, умерших от болезней (1); микроскопические методы изучения тканей (2); эксперимент, позволяющий моделировать на ...
... М., 1976; Система, структура и процесс развития международных отношений / Отв. ред. В.И. Ганпман. — М., 1984. 17. См., например: Антюхчна-Московченко В.И., Злобин А.А., Хруста-лев М.А. Основы теории международных отношений. — М., 1988, с. 68. 18. Возе К. 5осю1ое1е (1е 1а ра1х. — Рапа, 1965, р. 47—48. 19. ВгаШаг<1 РН., Д/аИН М.-К. Ьех ге1аиоп5 т1етайопа1е&. — Рапа, 1988, р. 65-71. 20. ...
0 комментариев