Навигация
Моногенные и полигенные заболевания
79253
знака
0
таблиц
0
изображений
2 Моногенные и полигенные заболевания
Моногенные заболевания обусловлены мутациями или отсутствием отдельного гена. Мутации могут захватывать один или оба аллеля. Клинические проявления возникают в результате отсутствия генетической информации или реализации дефектной. Моногенные заболевания исследуются в полном соответствии с законами Менделя (аутосомное или сцепленное с Х-хромосомой). Известно около 5000 моногенных заболеваний, более половины наследуется по аутосомно-доминантному типу.
К этой группе заболеваний относятся:
— неврофиброматоз (болезнь Реклингхаузена), при котором наиболее тяжело поражается нервная система;
— миотоническая дистрофия с миотонией, мышечной слабостью, катарактой, сердечной аритмией, нарушенной толерантностью к глюкозе, умственной отсталостью;
— синдром Марфана — наследственная болезнь соединительной ткани. Наиболее специфическими признаками являются нарушения скелета, вывих хрусталика, сердечнососудистые изменения, эктазия твердой мозговой оболочки;
— синдром Элерса - Данло — врожденная гиперрастяжимость соединительной ткани в связи с нарушением синтеза коллагена, обусловленным мутациями в разных коллагеновых генах;
— фенилкетонурия, связанная с недостаточностью печеночного фермента фенилаланингидроксилазы, локус которой расположен в длинном плече хромосомы 12. Дети с фенилкетонурией рождаются здоровыми, но в первые, же недели после рождения в связи с поступлением фенилаланина в организм с молоком матери развиваются клинические проявления заболевания: повышенная возбудимость, гиперрефлексия, повышенный тонус мышц, судорожные эпилептиформные припадки; от ребенка исходит «мышиный» запах. Позже развиваются умственная отсталость, микроцефалия;
— муковисцидоз (кистозный фиброз), в основе которого лежит нарушение транспорта ионов хлора и натрия через клеточные мембраны (ген муковисцидоза локализован в хромосоме 7), что приводит к избыточному выведению хлоридов. Отмечается гиперсекреция густой слизи в клетках эндокринной части поджелудочной железы, эпителии бронхов, слизистой оболочке желудочно-кишечного тракта;
— адреногенитальный синдром (врожденная гиперплазия коры надпочечников) относится к группе наследственных нарушений синтеза стероидных гормонов. Наиболее распространенная форма врожденной гиперплазии коры надпочечников — дефицит 21-гидроксилазы, ген локализован в коротком плече хромосомы 6;
— миопатия Дюшенна, вызванная мутацией в гене, ответственном за синтез белка дистрофина (ген расположен в локусе X^21). Заболевание проявляется прогрессирующей мышечной слабостью, дистрофией и некрозом отдельных мышечных волокон;
— гемофилия А — заболевание, сцепленное с Х-хромосомой, ген расположен в локусе Х28, мутация гена обусловливает дефицит фактора VIII. Клинические проявления состоят в нарушении гемостаза, увеличении времени свертывания.
Полигенные болезни обусловлены взаимодействием определенных комбинаций аллелей разных локусов и экзогенных факторов. Заболевания контролируются сразу несколькими генами, не подчиняются законам Менделя и не соответствуют классическим типам аутосомно-доминантного, аутосомно-рецессивного наследования и наследования, сцепленного с Х-хромосомой. Проявление признака во многом зависит от экзогенных факторов. Генетический риск полигенных болезней в большой степени зависит от семейной предрасположенности и от тяжести заболевания у родителей. Генетический риск полигенных болезней рассчитывают с помощью таблиц эмпирического риска. Определить прогноз нередко сложно.
Синдром Марфана.
В качестве примера клиники и генетики генных болезней рассмотрим некоторые нозологические формы более подробно.
Синдром Марфана - наследственная доминантная болезнь соединительной ткани. Клиническая идентификация синдрома была сделана В. Марфаном в 1886 году. Причиной синдрома Марфана являются мутации в гене фибриллина, ведущие к нарушению его синтеза. Обнаружение этих генных нарушений дает возможность проводить молекулярно-генетическую диагностику, включая пренатальную. Симптоматика синдрома Марфана разнообразна с многосистемностью поражений. Клинический полиморфизм по тяжести течения выражен значительно: от легких форм, трудно отличимых от нормы, до инвалидизирующего течения. Наиболее специфическими для синдрома Марфана являются нарушения скелета, вывих хрусталика, изменения в сердечнососудистой системе, эктазия твердой мозговой оболочки.
Характерные поражения:
Для поражения опорно-двигательной характерно – арахнодактилия; высокий рост, длинные конечности; деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз); деформация грудной стенки(вдавленная или «куриная» грудь);ненормальная подвижность суставов(гиперподвижность или контрактура); плоская стопа; высокое арковидное небо; недоразвитость верхней вертлужной впадины или мышечная Поражения глаз, кроме вывиха хрусталика, проявляются также в виде, миопии, большой и уплощенной роговице. При исследовании сердечнососудистой системы выявляются аортальная и митральная регургитация, пролапс митрального клапана, аневризма восходящей части аорты и расслоение аорты. При исследовании нервной системы - уже упомянутая, эктазия твердой мозговой оболочки, включая пояснично-крестцовое менингоцеле, и другие аномалии развития. При несомненном наличии болезни у родственников первой степени родства диагноз можно поставить, если у пациента в двух и более системах есть проявления болезни, включая специфические (вывих хрусталика, расширение и расслоение аорты, эктазия твердой мозговой оболочки). При несомненном отсутствии больных родственников первой степени родства диагноз ставится при условии обнаружения нарушений скелета и вовлечения в патологический процесс еще, по меньшей мере, двух других систем, включая одну с наиболее специфическими проявлениями. Частота синдрома Марфана в популяции равна 1:10000- 1:15000. Популяционных и этнических отличий в частоте и клинической картине болезни не отмечено. Синдром Марфана - типичная аутосомно-доминантная болезнь, хорошо изученная в клинико-генетическом плане. Клинический полиморфизм выражен очень ярко, но причины его неясны. С увеличением возраста отца (особенно после 35 лет) вероятность рождения ребенка с синдромом Марфана повышается.
Фенилкетонурия.
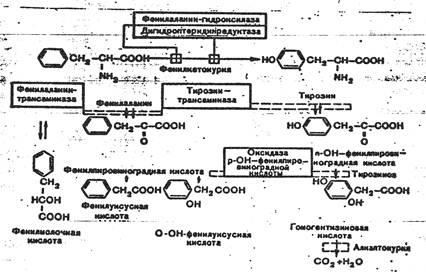
Фенилкетонурия - аутосомно-рецессивная болезнь аминокислотного обмена. Клинически фенилкетонурия выделена в самостоятельную форму в 1934 г. А. Фелингом. Патологические проявления связаны с недостаточностью печеночного фермента фенилаланингидроксилазы.
Этиология. Патогенез.
Недостаточность фермента ведет к нарушению процесса гидроксилирования фенилаланина в тирозин. Следствия этого - накопление фенилаланина в крови в больших концентрациях (фенилаланинемия), образование избыточного количества фенилпировиноградной, фенилуксусной и фенилмолочной кислот и нарушение формирования миелиновой оболочки вокруг аксона в ЦНС. Кроме того, высокая концентрация фенилаланина оказывает ингибирующее влияние на ряд ферментных систем, участвующих в метаболизме других аминокислот. Дети с фенилкетонурией рождаются здоровыми, но в первые месяцы жизни в связи с поступлением фенилаланина в организм с молоком матери развиваются клинические проявления: повышенная возбудимость, гиперрефлексия, повышенный тонус мышц, тремор, судорожные эпилептиформные припадки, характерный "мышиный" запах. Позже развиваются умственная отсталость, микроцефалия. Поскольку нарушение обмена фенилаланина ведет к снижению уровня тирозина, одно из проявлений заболевания - снижение уровня или прекращение образования меланина, поэтому у больных отмечается уменьшенная пигментация кожных покровов, волос, радужной оболочки глаз. Течение болезни прогредиентное. При отсутствии лечения умственная отсталость может достигать тяжелой степени. Диагноз ставится на основании клинической картины и биохимического исследования мочи (обнаружение фенилпировиноградной кислоты) и крови (гиперфенилаланинемия).
Ранняя диагностика фенилкетонурии и профилактическое лечение (диета) предупреждают развитие клинической картины болезни. Генетика фенилкетонурии хорошо изучена. Уже через год после клинического описания болезни Л. Пенроуз доказал аутосомно-рецессивный характер наследования. Ген фенилкетонурии (фенилаланингидроксилазы) расположен в длинном плече двенадцатой хромосомы. Для большинства семей возможны молекулярно-генетическая пренатальная диагностика и выявление гетерозигот. Популяционная генетика фенилкетонурии, как и большинства аутосомно-рецессивных болезней сложная. Частота заболевания в европейских странах в среднем составляет 1:10000 новорожденных, а частота гетерозигот 1:100.
СиндромДауна.
Рассмотрим некоторые хромосомные болезни. Синдром Дауна, трисомия по 21-й хромосоме - самая частая и наиболее хорошо изученная хромосомная болезнь. Частота рождения детей с синдромом Дауна составляет примерно 1:750 и не имеет какой-либо временной, этнической или географической разницы и родителей одинакового возраста. С возрастом (в большей степени матери и в меньшей мере отца) вероятность рождения ребенка с данной патологией существенно возрастает, и в возрасте 45 лет составляет около 3 %. Цитогенетические варианты синдрома Дауна разнообразны. Основную долю составляют случаи полной трисомии 21 как следствие нерасхождения хромосом в мейозе. Наряду с этим известны случаи регулярной трисомии, связанной с транслокацией 21-й хромосомы на другую - 21, 22, 13, 14 или 15-ю хромосому. Почти 50 % транслокационных форм наследуется от родителей носителей и 50 % - вновь возникшие мутации. Соотношение мальчиков и девочек среди новорожденных с синдромом Дауна составляет 1:1. Клиническая картина синдрома Дауна разнообразна: врожденные пороки развития, нарушения постнатального развития нервной системы, иммунодефициты и другие отклонения. Многие симптомы заметны уже при рождении ребенка и дальнейшем проявляются еще более отчетливо. Из черепно-лицевых дизморфий отмечается монголоидный разрез глаз, круглое уплощенное лицо, плоская спинка носа, крупный язык, брахицефалия, деформированные ушные раковины. Так же характерны мышечная гипотония и разболтанность суставов.
Часто диагностируются врожденный порок сердца, клинодактилия. Встречаются изменения дерматоглифики в виде четырехпальцевой, или "обезьяньей", складки на ладони, две кожные складки вместо трех на мизинце. Характерен низкий рост (на 20 см ниже среднего). Диагноз синдрома Дауна ставится на основании клинически на основании сочетания ряда симптомов. Наиболее важные из которых: уплощение профиля лица (90 %), отсутствие сосательного рефлекса (85 %), избыток кожи на шее (80 %), монголоидный разрез глаз (80 %), мышечная гипотония (80 %), разболтанность суставов (80 %), диспластический таз (70 %), деформированные ушные раковины (40 %), клинодактилия мизинца (60 %), четырехпальцевая сгибательная складка (поперечная линия) на ладони (40 %). Большое значение для диагностики имеет задержка умственного и физического развития ребенка. Задержка умственного развития может достигать степени имбицильности, а коэффициент умственного развития у разных детей широко варьируется (IQ от 25 до 75). Больные с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела. Врожденные пороки внутренних органов и недостаточность иммунной системы часто приводят к летальному исходу в первые 5 лет жизни. Дифференциальная диагностика проводится с другими формами хромосомных аномалий и врожденным гипотиреозом. Цитогенетическое исследование показано и при подозрении на синдром Дауна и при клинически установленном диагнозе. В последнем случае это необходимо для прогноза здоровья будущих детей у родителей ребенка и их родственников. Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врожденные пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющая терапия, защита от действия вредных факторов внешней среды. Многие больные с трисомией 21 способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семью.
Синдром Патау
Синдром Патау - трисомия по 13-й хромосоме, выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования у детей с врожденными пороками развития. Обнаружены простые и транслокационные формы трисомии 13, однако клинически и патологоанатомически они неразличимы. Частота синдрома Патау среди новорожденных составляет 1:6000. Соотношение полов при данной патологии близко 1:1. Частое осложнение при вынашивании плода с синдромом Патау - многоводие (50 %). Для заболевания характерны множественные, тяжелые пороки развития головного мозга, мозговой и лицевой частей черепа, внутренних органов. Окружность черепа обычно уменьшена, лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположены и деформированы (80 %). Типичный признак - расщелина верхней губы и неба (70 %). Всегда обнаруживаются пороки внутренних органов в разных комбинациях: пороки сердца (80 %), незавершенный поворот кишечника (40 %), кисты почек (42 %), аномалии внутренних половых органов (73 %), дефекты поджелудочной железы (43 %). Часто наблюдается полидактилия кистей (50 %) и их флексорное положение(44 %). Дети с синдромом Патау практически всегда имеют глубокую идиотию. Клиническая диагностика основывается на сочетании характерных пороков развития. Однако решающий фактор в диагностике - исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших больных, с целью составления прогноза для будущих детей в семье. Лечебные мероприятия неспецифичны: общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни, но некоторые больные живут до нескольких лет.
Синдром Клайнфельтера.
Синдром Клайнфельтера относится к группе полисомий по половым хромосомам. Заболевание включает в себя случаи полисомии, при которых имеется не менее двух Х-хромосом и не менее одной Y-хромосомы. Наиболее часто (примерно 1:600) встречается синдром Клайнфельтера с набором 47,XXY.Этот синдром является и наиболее типичным клинически. Варианты полисомии с большим числом Х - и Y-хромосом (XXXY,XYY,XXXXY,XXYY) встречаются редко. Присутствие Y-хромосомы определяет формирование мужского пола. До периода полового созревания мальчики развиваются почти нормально. Вызываемый добавочной Х-хромосомой генетический дисбаланс проявляется клинически в период полового созревания в виде недоразвития семенников и вторичных мужских половых признаков. Мужчины с синдромом Клайнфельтера обычно имеют высокий рост, астеническое или евнухоидное телосложение, слабое оволосение лица, подмышечных впадин и лобка. Выявляется умственная отсталость легкой и средней степени, а в четверти случаев гинекомастия. Больные бесплодны (азооспермия, олигоспермия).
Синдром Шерешевского-Тернера.
Синдром Шерешевского-Тернера - единственная форма моносомии у живорожденных. Цитогенетика синдрома разнообразна. Более половины всех больных данным синдромом имеют простую полную моносомию по Х-хромосоме (45,Х). В остальных случаях наблюдаются мозаичные формы и более редкие формы со структурными аномалиями Х-хромосом (делеция, транслокация и другие аномалии). Клинически синдром Шерешевского-Тернера проявляется следующими признаками. Со стороны половой системы отмечается либо полное отсутствие гонад (агенезия), либо гипоплазия матки и маточных труб, первичная аменорея, недостаток эстрогенов, половой инфантилизм. Встречаются различные пороки сердечно-сосудистой системы и почек. Снижения интеллекта не отмечается, однако больные обнаруживают эмоциональную неустойчивость и инфантилизм психических процессов. Внешний вид больных своеобразен. Отмечаются характерные симптомы: короткая шея с избытком кожи и крыловидными складками; в подростковом возрасте выявляется отставание в росте и развитии вторичных половых признаков; для взрослых характерны нарушения скелета, черепно-лицевые дизморфии, вальгусная девиация коленных и локтевых суставов, низкое расположение ушных раковин, диспропорции тела (укорочение ног, относительно широкий плечевой пояс, узкий таз). Рост взрослых больных на 20-30 см ниже среднего. Лечение больных с синдромом Шерешевского-Тернера комплексное и включает в себя реконструктивную и пластическую хирургию, гормональную терапию (эстрогены, гормон роста), психотерапию.
Синдром кошачьего крика.
Синдром кошачьего крика - частичная моносомия по короткому плечу 5-й хромосомы (5p-). Синдром обусловлен делецией короткого плеча 5-й хромосомы. У детей с этой хромосомной аномалией отмечается необычный плач, напоминающий требовательное кошачье мяуканье или крик. Частота синдрома достаточно велика для делеционных синдромов - 1:45000. Цитогенетически в большинстве случаев выявляется делеция с утратой от трети до половины короткого плеча 5-й хромосомы, реже наблюдается полная утрата короткого плеча. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Клиническая картина синдрома довольно сильно варьируется у отдельных больных по сочетанию врожденных пороков развития органов. Наиболее характерный признак - "кошачий крик" - обусловлен изменением гортани. У большинства больных имеются те или иные изменения мозгового черепа и лица: лунообразное лицо, микроцефалия, микрогения, антимонголоидный разрез глаз, высокое небо, плоская спинка носа, деформация ушных раковин. Кроме того, встречаются врожденные пороки сердца, костно-мышечной системы и внутренних органов. Выраженность клинической симптоматики меняется с возрастом. "Кошачий крик", мышечная гипотония, лунообразность лица с возрастом исчезают, а микроцефалия выявляется более отчетливо, прогрессирует психомоторное недоразвитие, косоглазие. Продолжительность жизни больных зависит от выраженности клинической картины в целом, тяжести врожденных пороков внутренних органов (прежде всего сердца), уровня оказываемой медицинской помощи и повседневной жизни. Большинство больных умирает в первое десятилетие жизни. Во всех случаях больным и их родителям показано цитогенетическое обследование.
Похожие работы
... , но и в обычных условиях жизни у практически здоровых людей , уделяющих недостаточное внимание разнообразию пищевого рациона . Развитию этой формы витаминной недостаточности способствуют широкое использование в питании рафинированных продуктов , лишенных витаминов в процессе их производства ( хлеба тонкого помола , сахара и др.); потеря витаминов при длительном хранении и неправильной кулинарной ...
... разработке? ) генетической инженерии для лечения наследственных болезней, даже если будут сделаны решительные прорывы в синтезе соответствующих генов и способах их доставки в клетки-мишени. Генетика человека еще не располагает достаточными сведениями обо всех особенностях функционирования генетического аппарата человека. Пока еще неизвестно, как он будет работать после введения дополнительной ...
... , в известной степени, определяется генотипом. Кроме хромосомной, различают внехромосомную, или цитоплазматическую, наследственность. Хромосомная наследственность связана с распределением носителей наследственности (генов) в хромосомах. Цитоплазматическая наследственность проявляется в наследовании признаков, которые контролируются внехромосомными, цитоплазматическими наследственными факторами, ...
... Сфингомиелиназа Сфингомиелин, холестерин. Лейкоциты, материал биопсии органов, фибробласты кожи (инфантильная и ювенильная формы) Материал биопсии Гиперлипопротеидемии (ГЛП) Наследственные заболевания нарушений липидного обмена представлены большим количеством нозологических форм, которые обусловливают развитие атеросклероза и других форм сердечно-сосудистой патологии. Впервые ...
0 комментариев