Навигация
Молекулярні моногенні спадкові захворювання
31291
знак
0
таблиц
0
изображений
2.2 Молекулярні моногенні спадкові захворювання
Більшість спадкових моногенних захворювань — це дефекти обміну речовин. За класифікацією ВООЗ, спадкові дефекти обміну речовин поділяються на 11 груп. Це захворювання, зумовлені порушенням: 1) амінокислотного обміну;
2) вуглеводневого;
3) ліпідного;
4) стероїдного;
5) пуринового і піримідинового;
6) обміну речовин у сполучній тканині, кістках і м'язах;
7) структури гему і порфірину;
8) обміну речовин в еритроцитах і порушення їх структури.
Крім того, виділяють ще такі групи:
9) аномалії обміну металів;
10) захворювання, які характеризуються дефектом транспорту різних речовин; 11) захворювання, спричинені аномаліями будови і функції ферментів і білків плазми.
Тепер відомо більш як 1000 спадкових захворювань, зумовлених дефектом обміну речовин. Найбільшу групу становлять захворювання, спричинені порушенням обміну речовин, які представлені чотирма класами білків:
1) білками-ферментами;
2) структурними;
3) транспортними;
4) циркулюючими.
Дефекти обміну структурних і циркулюючих білків можна виявити, вивчаючи будову цих білків (аномальні гемоглобіни, псевдохолінестерази). Ці дефекти обміну білків-ферментів і циркулюючих білків визначаються концентрацією продуктів метаболізму, який здійснюється даним ферментом у крові і сечі хворого.
Більшість спадкових дефектів обміну — це ферментопатії, тобто порушення будови білків-ферментів, які беруть участь в обміні тих чи інших речовин. При цьому в організмі спостерігається дефіцит кінцевого продукту обміну і накопичення проміжних продуктів (дериватів або мінорних речовин).
Добре вивченими молекулярними хворобами людини є різні гемоглобінопатії та фенілкетонурія. Гемоглобінопатії найчастіше — це наслідок порушення структури і регуляції синтезу глобінів, а фенілкетонурія — порушення структури одного з ферментів, які регулюють обмін фенілаланіну і перетворення його через ряд етапів до тирозину і меланіну.
На прикладі гемоглобінопатій ми бачимо, що молекулярна хвороба виникає через порушення функції структурного білка.
Проте молекулярні хвороби можуть виникати і через порушення функції білка-ферменту, внаслідок чого порушується обмін тієї чи іншої речовини. За таким типом розвивається більшість хвороб обміну речовин. Розглянемо, це на прикладі фенілкетонурії та інших хвороб.
Фенілкетонурія відноситься до аутосомнорецесивних захворювань. Близько до фенілкетонурії стоїть ряд інших захворювань, зумовлених порушенням обміну фенілаланіну. Для наочності наведемо більш детальнішу схему обміну фенілаланіну; і йог» порушень.
Одним із продуктів обміну фенілаланіну є тироксин (гормон щитоподібної залози), який утворюється з тирозину. Цей гормон впливає на загальний обмін. При дефекті вказаного ферменту в ембріональному періоді онтогенезу у дітей розвивається незоровий кретинізм (особливий вид карликовості).
У хворих — атрофічна щитоподібна залоза. У фенотипі таких хворих відмічається вкорочення кінцівок при нормальному розмірі тулуба і голови. Допускається, що щитоподібна залоза у цих хворих в ембріональному періоді була зруйнована материнськими антитілами проти щитоподібної залози. Незобовий кретинізм виникає після другої вагітності матері, вік якої понад ЗО років. Відомо, що зі збільшенням віку матері концентрація антитіл до білків щитоподібної залози наростає. Захворювання успадковується аутосомно-рецесивно.
До спадкових захворювань порушення вуглеводневого обміну належить глікогеноз, галактоземія та інші захворювання.
Галактоземія.
Галактоза — це складова частина молочного цукру (лактози). В організмі вона за допомогою ферменту галактозо-1-фосфат-уридилтрансферази перетворюється в галактозо-1-фосфат, який далі перетворюється в глюкозо-6-фосфат, що входить до метаболічного циклу глюкози.
При галактоземії у дитини відзначається недостатність вказаного ферменту, відбувається накопичення в організмі галакто-зо-1-фосфату, який впливає токсично на тканини організму. У дитини розвивається цироз печінки, уражуються нирки, внаслідок чого вони не здатні реабсорбувати амінокислоти з провізорної сечі. Це призводить до аміноацидурії. З галактози утворюється дульцитол, який накопичується в кристалику ока і призводить до ранньої катаракти.
У дітей через не переносність материнського молока спостерігаються диспепсичні розлади, вони худнуть, з'являється жовтяниця, затримується психічний розвиток. Ці діти вмирають у перші місяці життя, якщо не буде своєчасно призначене необхідне лікування. Якщо дитина залишається живою, то у неї відзначається мікроцефалія, знижується тонус м'язів, з'являються судоми, гепато і спленомегалія, розвивається анемія.
За своєчасної діагностики захворювання, відлучення дитини від груді і переведення на годування коров'ячим молоком (в якому галактози менше, ніж у жіночому), а також на спеціальну дієту можна домогтися значного терапевтичного ефекту. Рання діагностика захворювання проводиться на ауксотрофних мікробах, а також хроматографічним визначенням концентрації амінокислот у крові дитини. Захворювання успадковується аутосомно-рецесивно.
Близьким до порушення вуглеводневого обміну є мукополісахаридози.
Глікозамінглікани (мукополісахариди) — це аміноцукор (полісахарид) у комплексі з глюкуроновою, сіаловою, сірчаною і оцтовою кислотами. Аміноцу-кор — це глюкоза, в якій гідроксильна група (ОН) при другому атомі вуглецю замінена на аміногрупу (МН2). Глікозамінглікани в різних органах відрізняються складом амінокислот і наявністю сульфгідрильних груп. Відомо 9 типів глікозамінгліканів: хондроітинсульфат, гепаринсульфат, кератинсульфат, гіалу-ронова кислота тощо.
При порушенні синтезу і деградації глікозамінгліканів вони відкладаються в клітинах організму і розвивається мукополісахаридоз. Захворювання вперше було описано Гурлером у 1919 р. і було визначене як гаргоїлізм, тому що голова хворих нагадує гаргоїлів — потворних фігур на паризькому соборі. В наступні роки було встановлено, що це збірна група захворювань, при яких порушується обмін одного з гліканів.
Одним із мукополісахаридозів є синдром Гурлера. У хворих через дефект ферменту а - ідуронідази відбувається накопичення гепарину і дерматансульфату. Захворювання звичайно діагностується на другому році життя дитини. Воно виявляється розумовою відсталістю і скелетними змінами: кіфозом, випинанням лоба, плоским носом, потовщенням губ, дрібними зубами, збільшеним язиком, деформованими вушними раковинами, густим жорстким волоссям, коротким тулубом, деформованою грудною кліткою, збільшенням поперекового і грудного кіфозу, потовщеними і розширеними епіфізами довгих кісток. Пальці рук при синдромі Гурлера набувають напівзігнутого положення, відзначається деяка скутість хворих. У них збільшений живіт, часті пупкові грижі, гепатоспленомегалія. Спостерігаються серцеві вади, помутніння рогівки, нерідко — вроджена глаукома.
Захворювання діагностується шляхом виявлення в сечі підвищеної концентрації кислих глікозамінгліканів (гепаринсульфату). В культурі фібробластів у лейкоцитах відзначається зниження концентрації а-1-ідуроиідази. Успадковується захворювання аутосомно-рецесивно.
Більш легкою формою мукополісахаридозу є синдром Шейе, зумовлений дефектом того самого ферменту. Ці хворі живуть довго.
Пізніше були описані інші синдроми, клінічно подібні до синдрому Гурлера, але з дефектом інших ферментів обміну глікозамінгліканів. Це синдроми Санфіліппо, Моркіо, Марото — Ламі, Гунтера. Всі вони успадковуються аутосомно-рецесивно. Один з них — синдром Гунтера — успадковується рецесивне, зчеплено зі статевою хромосомою, хворіють хлопчики. Це захворювання — генокопія синдрому Гурлера, через те що у хворих є багато спільних симптомів. Захворювання протікає більш м'яко.
До спадкових порушень обміну ліпідів належать хвороби Тея Сакса, Німана — Піка, Гоше. Це — дізосомні захворювання. Через дефекти лізосомних ферментів, які беруть участь у метаболізмі ліпідів, останні накопичуються всередині клітин і викликають їхню загибель.
Не дивлячись на рідкість цих хвороб і невелике продовження життя хворих, внутрішньоклітинні ліпідози з біохімічної сторони вивчені більш детально, ніж інші спадкові захворювання нервової системи, завдяки вивченню культури клітин і біопсійного матеріалу.
Ліпіди — це складні сполуки ненасиченого аміноспирту сфінгозину, жирних кислот і вуглеводів. Найпростішим ліпідом є церамід (сполука сфінгозіну з жирною кислотою). При наступному сполученні цераміду з глюкозою, галактозою та іншими речовинами утворюються більш складні ліпіди: сфінгоміелін, галактоцереброзид, глюкоцереброзид, гангліозид тощо. Перетворення одного ліпіду в інший здійснюється за допомогою відповідного ферменту. Дефект того чи іншого ферменту призводить до накопичення всередині клітин відповідного ліпіду, що називається ліпідозом, або сфінголіпідозом.
При хворобі Тея Сакса здійснюється накопичення всередині клітин гангліозиду (сполука цераміду з глюкозою, галактозою і М ацетилнейраміновою кислотою). Захворювання розвивається на першому півріччі життя дитини і виявляється прогресуючою психічною деградацією і сліпотою.
При хворобі Німана — Піка в клітинах головного мозку і внутрішніх органах накопичується сфінгомієлін (сполука цераміду з холіном і фосфорною кислотою). В одних випадках захворювання розвивається в дитячому віці, а в других — у зрілому й похилому. Виявляється органічною неврологічною симптоматикою (інфантильністю) і спленомегалією.
При хворобі Гоше в клітинах головного мозку і внутрішніх органах накопичується глюкоцереброзид (сполука цераміду з глюкозою). Виділяють гостру, підгостру і хронічну форми захворювання.
При гострих формах захворювання спостерігається м'язова ригідність, порушення зору, психічна деградація, остеопороз кісток, стоншення кортикального шару їх, деформація стегон за типом «пляшок» або колб Ерленмейера. Часті переломи кісток через розростання клітин Гоше в кістковому мозку. Зменшення кісткового мозку призводить до гіпохромної анемії, тромбопенії, що супроводиться носовими та іншими кровотечами. Через гепатоспленомегалію у дітей різко збільшений живіт. На очному дні так само, як і при хворобі Німана — Піка, виявляється вишнево - червона пляма. Такі діти звичайно вмирають на першому році життя від виснаження.
При під гострій формі захворювання всі симптоми мають більш м'який характер.
При хронічній (вісцеральній) формі захворювання пошкоджуються внутрішні органи без втягнення в процес головного мозку. Хвороба Гоше — це гетерогенне захворювання. Описані як аутосомно-рецесивні, так і аутосомно-домінантні форми успадкування.
Клінічно ранні форми хвороби Німана — Піна і хвороби Гоше мають багато спільних симптомів, тему диференціальна діагностика їх проводиться на підставі дослідження біопсійного матеріалу (клітини печінки).
Близько половини білків, у тому числі і ферментів, складаються із кількох (не менше двох) поліпептидів, кожний з яких синтезується під контролем одного з двох алельних генів гомологічних хромосом. Для розвитку виражених ензимопатій потрібна наявність дефекту в обох алелях гена, через те дефект тільки в одному алелі може клінічно не виявитись, тому що синтезується один нормальний поліпептид. Отже, ензимопатії — це найчастіше аутосомно-рецесивні захворювання. Вони розвиваються у гомозигот за патологічним геном. Гетерозиготних носіїв патологічного гена можна виявити навантаженням певними хімічними речовинами або іншими методами біологічних досліджень.
Ферменти, які складаються з кількох поліпептидів, кодуються різними генними локусами, причому в різних тканинах організму вони мають різну активність. Існує декілька ізоферментів лактатдегідрогенази (ферменту, який каталізує взаємне перетворення піровиноградної кислоти в молочну і навпаки), декілька ферментів фосфоглюкомутази, яка каталізує взаємне перетворення глюкозо-1-фосфату в глюкозо-6-фосфат, кілька ферментів, які беруть участь в обміні глікогену тощо.
При хворобі Герса — це один із типів глікогенозу — порушений розпад печінкового глікогену, а м'язовий розпадається нормально, .хоча в обох випадках дефектним ферментом є фосфорилаза глікогену. Різні ферменти мають свій оптимум активності при відповідному рН середовища. Цим пояснюється різноманітність спадкових порушень обміну речовин при одній і тій самій формі ензимопатій.
Відомі й інші механізми розвитку спадкових молекулярних захворювань. При дії на клітини організму ультрафіолетових променів іонізуючої радіації або хімічних речовин в молекулі ДНК можуть утворюватися зв'язки між двома азотистими основами (утворюються димеритиміна та інших азотистих основ). У нормі ці порушення усуваються репаративними системами клітин. Проте, при неповноцінності репаративних систем указані димери не усуваються. Чим більше димерів піримідинів утворюється в клітині, тим менш життєздатною вона стає, а це сприяє виявленню багатьох хвороб (так звані хвороби репарації ДНК). До таких захворювань відносяться пігментна ксеродерма, анемія Фанконі, прогерія, синдром Блюма, атаксія телеангіектазія та ін.
Встановлено, що ДНК організму людини містить до 3 млрд. нуклеотидів. Під час поділу клітини кількість помилок нуклеотидної послідовності може бути до 50 тис. У клітинах людського організму при температурі 37 °С протягом одного дня втрачається спонтанно 20 тис. азотистих основ. Зовнішні впливи збільшують число цих втрат.
Так, під впливом ультрафіолетового опромінення виникають зміни в молекулі ДНК у вигляді утворення міцних хімічних зв'язків між піримідиновими азотистими основами (Т, Ц) одного ланцюга. При цьому порушуються водневі зв'язки між двома ланцюгами ДНК і утруднюється реплікація ланцюгів. Під час опромінювання рентгенівськими або гамма-променями відбувається розрив подвійного ланцюга ДНК головним чином за рахунок продуктів радіолізу води до водню і гідроксилу (Н і ОН).
Хімічні речовини (як самі, так і продукти їх перетворення) міцно зв'язуються з пуриновими азотистими « основами ДНК (аденін, гуанін) і таким чином можуть з модифікувати зазначені основи. До таких речовин відноситься і бензошрен, який знаходиться в тютюновому димі цигарок.
Інші мутагени послаблюють зв'язку пуринових основ з дезоксирибозою і вони легко втратяться. У нормі ці дефекти усуваються репаративними с системами.
Є декілька способів репарації.
Один з них темповий ферментативний. Фермент ендонуклеаза знаходить місце дефекту і розриває біля нього ланцюг ДНК.
Другий фермент екзонуклеаза розриває цю ділянку з іншого боку і усуває його. Утворений отвір одному ланцюзі подвійної спіралі за допомогою ферменту ДНК- полімерази заповнюється нуклеотидами які комплементарні нуклеотидами позачіпленого ланцюга.
Потім фермент лігаза знову утворений фрагмент з останнім ланцюгом і цілість подвійного ланцюга ДНК відновлюється. Якщо отвір дуже великий ферментна система не в змозі зсунути цей дефект, тоді в дію вступає інший механізм репарації ДНК ( постреплікаційна, 503-репарація тощо).
Під час реплікації молекула ДНК розділяється на одиночні ланцюги і на цих ланцюгах реплікація виникає за допомогою ДНК- полімерази за рахунок вільних нуклеотидів. Проте нуклеотиду можуть бути не комплементарними, а випадковими. Тоді виникає мутація, яка буде .передаватись наступними дочірніми клітинами під час їхнього поділу. Чим більше невиправлених дефектів утворюється в клітині, тим менш життєздатною стає клітина. Це прискорює її старіння і спричинює виникнення ряду хвороб репарації. У процесі репарації можуть виникати помилки відновлення.
Репарації — могутнє джерело мутацій в організмі. Відомо близько 20 хвороб, зумовлених порушенням репарації ДНК і багато молекулярних спадкових захворювань, зв'язаних з порушенням реплікації ДНК. Наведемо ще одну групу спадкових молекулярних генетичних захворювань. Це спадкові порушення обміну білірубіну (спадкові пігментні гепатози) — відносно доброякісні жовтяниці, які зумовлені порушенням обміну білірубіну — продукту перетворення гемоглобіну.
Гемоглобін відмерлих еритроцитів у клітинах системи мононуклеарних фагоцитів розпадається до глобіну, залізовмісного гемосидерину і без залізовмісного гематоїдину. Глобін розкладається до амінокислот, які йдуть на побудову білків організму. Залізо піддається окисленню й використовується організмом у вигляді феритину.
Гематоїдин перетворюється в білірубін. Білірубін поступає в кров'яне русло, звідки захвачується клітинами печінки, де відбувається з'єднання його з глюкуроновою кислотою за допомогою ферменту глюкуроніл трансферази. Білірубін глюкуронід попадає разом з жовчю в кишки, де перетворюється в уробіліноген і уробілін.
Вільний білірубін у воді не розчинний, його можна виявити в сироватці крові непрямою реакцією Гіманс-ван-ден-Берга. Білірубін у сполуці з глюкуроновою кислотою (білірубін-глюкуронід) у воді розчинний і його можна виявити прямою реакцією.
Підвищений вміст білірубіну в сироватці крові виявляється жовтяницею. Виділяють 3 види жовтяниць:
1. Передпечінкова жовтяниця, що виникає при підвищеному руйнуванні еритроцитів (розпад еритроцитів при малярії, гемолітичній анемії, інфаркті легень, великих гематомах, серпоподібно-клітинній анемії). При цих захворюваннях виникає велика кількість білірубіну, який не взмозі весь з'єднатися з глюкуроновою кислотою і в сироватці крові визначається вільний білірубін, який у воді не розчинний. Такий білірубін в сечу не поступає, тому реакція сечі на білірубін — негативна.
Похожие работы
... іциту альфа1-антитрипсина. Ідентифікація всіх генів людини, відкриття за допомогою біоінформатики нових генних мереж, незмірно збільшить можливості генетичного тестування спадкової схильності і медико-генетичного консультування. 4. Методи генетичного тестування Для клінічних цілей використовують різні методи генетичного тестування. «Генетичне тестування — аналіз людської ДНК, РНК, хромосом, ...
... синтезу борфтористих сполук (BF4, HBF4,). Солі важких металів з цими сполуками є добре розчинними у воді. BF4 важче повітря, і, ймовірно, за рахунок цього у місті загинули таргани, птахи, що проживали на нижньому ярусі. Канцерогенні фактори навколишнього середовища Онкологічні захворювання займають одне з перших місць серед причин захворюваності та смертності населення. Якщо у 1850 р. у ...

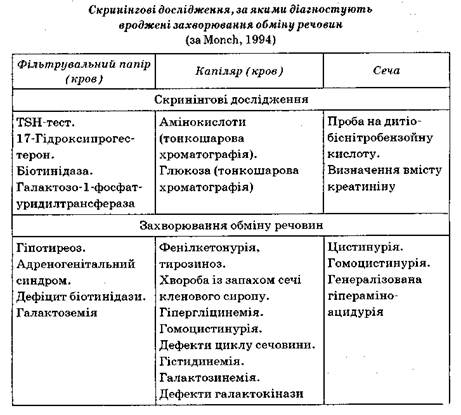
... нервово-психічному розвитку. Розумове відставання характеризується імбецильністю різних ступенів. У 40—50 % хворих виявляють вроджені вади серця і судин та інші аномалії. Лікування симптоматичне. 2 Спадкові хвороби обміну речовин Основні клінічні ознаки вроджених порушень метаболізму Рецидивуючі напади блювання, ацидоз (після початку годування грудним молоком або молочними сумішами) ...
... хворобі Дауна. У 1969 р. Т.Каперсон запропонував диференціальне фарбування хромосом , що дало змогу розрізняти кожну з хромосом окремо і виявляти зміни їх. Великий внесок у вивчення загальної генетики людини зробили: М.П.Дубинін , Д.Д.Ромашов , А.А.Малиновський , В.П.Єфроїмсон , М.П.Бочков , І.Р.Барляк , М.А.Пілінський. В Україні питаннями медичної генетики займалися такі відомі вчені , як ...
0 комментариев