Навигация
Получение ацетилферроцена
28638
знаков
0
таблиц
9
изображений
3.1. Получение ацетилферроцена
В трёхгорлую колбу ёмкостью 200 мл, снабжённую термометром, механической мешалкой и обратным холодильником, помещали 20 мл уксусного ангидрида и 0.8 мл концентрированной фосфорной кислоты и перемешивали 10 минут. Затем добавляли 3 г (0.016 моль) ферроцена, нагревали на песчаной бане до 110-113°С в течение 40 минут.
После охлаждения до комнатной температуры в реакционную смесь добавляли насыщенный раствор соды до полной её нейтрализации. После прекращения выделения углекислого газа, выпавший из раствора осадок фильтровали на воронке Бюхнера, тщательно промывали водой до нейтральной реакции промывных вод и высушивали на воздухе. Затем осадок растирали в ступке с 9 см3 окиси алюминия до порошкообразного состояния и помещали на слой чистой окиси алюминия в хроматографическую колонку (23х1.8 см). Петролейным эфиром вымывали непрореагировавший ферроцен, смесью петролейного и диэтилового эфиров (1:1) – ацетилферроцен. Далее смесь эфиров отгоняли вакуумным насосом (колба при этом находилась в горячей воде). Выкристаллизовавшееся вещество досушивали на воздухе.
Для определения химического состава полученного вещества были использованы метод тонкослойной хроматографии со свидетелем на окиси алюминия (II степени по Брокману), метод термо-визуального определения температуры плавления и метод протонного магнитного резонанса (в четырёххлористом углероде).
В ходе синтеза был получен ацетилферроцен массой 2.29 г (0.01 моль) с температурой плавления 86°С и выходом 62.50%.
ПМР-спектр 1H: 2.53 (C, 3H, COCH3); 4.12 (C, 5H, H1’); 4.34 (C, 2H, H3); 4.65 (C, 2H, H2).
3.2. Разделение ферроцена и ацетилферроцена
К 100 мл эфирного раствора смеси 0.37 г (2 ммоля) ферроцена и 0.46 (2 ммоля) ацетилферроцена добавляли 50 мл 5% раствор хлорида железа (III). Смесь перемешивали магнитной мешалкой при 20° С в течении 20 минут. Водный слой отделяли от эфирного. Затем эфирный раствор промывали водой три раза и сушили над безводным сульфатом натрия. Растворитель отгоняли, и вещество оставляли сушиться на воздухе.
Водный, зелёного цвета, раствор соли ферроцения обрабатывали водным 5% раствором тиосульфата натрия. Выпавший из раствора ферроцен экстрагировали эфиром. Эфирный раствор два раза промывали водой, сушили над безводным сульфатом натрия и отгоняли растворитель.
Для определения химического состава полученных веществ были использованы метод тонкослойной хроматографии со свидетелем на окиси алюминия (II степени по Брокману), и метод термо-визуального определения температуры плавления (для ферроцена проводится в запаянном капилляре).
Из смеси было выделено 0.44 г (1.92 ммоля) ацетилферроцена с температурой плавления 86°С (выход 96.07%) и 0.34 г (1.82 ммоль) ферроцена с температурой плавления 173°С (выход 91.40%).
4. Обсуждение результатов
4.1 Синтез ацетилферроцена
Синтез ацетилферроцена был проведён по следующей схеме:

При рассмотрении литературы, связанной с синтезом ацетилферроцена было найдено множество методик его получения (см. Литературный обзор).
Хлорид алюминия (III) применяется наиболее часто в качестве катализатора при ацилировании ферроцена. Однако он катализирует также и ряд других превращений ферроценового ядра. Так, при действии на ферроцен хлорангидридов карбоновых кислот, имеющих электроноакцепторные заместители, происходит окисление ферроцена, а ацилферроцен образуется с небольшим выходом или не получается вовсе. Кроме того, хлористый алюминий катализирует реакции с разрывом связи железо-кольцо (замена циклопентадиенального кольца на ареновое, частичная деструкция ферроценового ядра). Поэтому, мы применяли наиболее доступный в условиях лаборатории мягкий катализатор – концентрированную фосфорную кислоту.
В ходе синтеза очень важно было поддерживать температуру реакционной смеси в интервале от 110 до 113°С, т.к. при более низкой температуре возвращается много непрореагировавшего ферроцена, а при повышении температуры образуются смолообразные продукты.
Для полной нейтрализации фосфорной и непрореагировавшей уксусной кислот реакционную смесь оставляли на ночь с насыщенным раствором соды.
При разделении выпавшего осадка на хроматографической колонке часть оставшегося после реакции ферроцена окислилась на окиси алюминия, о чём свидетельствовал зеленоватый цвет использованной окиси. В ходе вымывания моноацетилферроцена был обнаружен диацетилферроцен по появившемуся на окиси алюминия фронту розового цвета. Однако нужный нам ацетилферроцен им загрязнен не оказался по данным полученной позже тонкослойной хроматограммы. Предположительно, для вымывания диацетилферроцена понадобился бы петролейный эфир.
4.2 Сравнение ОВ способностей ферроцена и ацетилферроцена
В просмотренной литературе (см. Литературный обзор) было найдено множество окисляющих реагентов и способов окисления ферроцена до ферроцений-катиона. Нами был выбран в качестве окислителя хлорид железа (III) как самый удобный реагент, т.к. соль тетрахлорферрата ферроцения является одной из наиболее устойчивых солей этого соединения. Окисление было проведено по схеме:
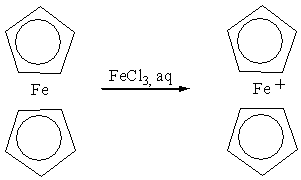
Хлористое олово было взято в избытке для максимально выхода ферроценй-катиона. После окисления ферроцений перешёл в водный раствор, о чём можно судить по синевато-зелёной окраске водного слоя после отделения его от эфирного.
Так как электроноакцепторные заместители в кольцах ферроцена затрудняют его окисление, ацетилферроцен не был окислен в тех же условиях, что незамещённый ферроцен:

Для окисления ацетилферроцена понадобились бы более жёсткие окислители, такие как AgBF4 и HAuCl4. Так как ферроцен и его производные не растворяются в воде, неокисленный ацетилферроцен остался в эфирном слое реакционной смеси, окрашивая его в насыщенный рыжий цвет. Не 100% выход ацетилферроцена после разделения обусловлен обычными для лабораторных условий потерями.
Для восстановления ферроцений-катиона мы воспользовались тиосульфатом натрия как доступным и эффективным реагентом. Мы не использовали органические восстановители из-за сложностей их разделения с восстановленным ферроценом. Реакция проходила по схеме:

Потери в ходе опыта, возможно, обусловлено сольватическим разложением соли ферроцения, несмотря на то, что водные растворы наиболее устойчивы.
В данной работе перед нами не стояло задачи выделения комплексной кристаллической соли ферроцений-катиона. Однако это можно было бы осуществить по схеме:
(C5H5)2+FeCl4- + NaB(C6H5)4aq ® [(C5H5)2]+ (C6H5)4- ¯ + Na[FeCl4]
В ходе реакции комплексная соль ферроцения образуется количественно.
4.3. Разделение ферроцена и его производных
Анализируя результаты, полученные в ходе нашей работы, можно предложить новый способ разделения ферроцена и его производных с электроноакцепторными заместителями в циклопентадиенальных кольцах. При синтезе ферроцениевых производных часть ферроцена всегда остаётся непрореагировавшим. Обычно продукт отделяют от исходного реагента на хроматографической колонке. Однако это не всегда возможно в условиях той или иной лаборатории. Предложенный нами метод очнь прост и эффективен

5. Выводы
1. В работе синтезирован ацетилферроцен (ацетил-дициклопентадиенилжелезо), проведена его очистка и идентификация.
2. Проведено исследование окислительно-восстановительных свойств ферроцена и моноацетилферроцена.
3. На основе проведённой работы предложен метод разделения ферроцена и его производных с электроноакцепторными заместителями в циклопентадиенальных кольцах.
6. Литература
1. Wilkinson G., Rosenblum M., Whiting M.C., Woodward R.B. - J. Amer. Chem. Soc., 1952, vol.74, p.2125
2. Bagus P.S., Walgren U.I., Almof J. – J. Chem. Phys., 1976, vol.64, p.2324
3. Eiland P.F., Pepinsky R. – J. Amer. Chem. Soc., 1952, vol.74, p.4971
4. Bohn R.K., Haaland A. – J. Organomet. Chem., 1966, vol.5, p.470
5. Mammato N.J., Zalkin A., Landers A., Rheingold A.L. – Inorg. Chem., 1977, vol.16, p.297
6. Rozenblum M., Santer J.O., Howells V.G. – J. Amer. Chem. Soc., 1963, vol.85, p.1450
7. Rozenblum M., Abbate F.V. – J. Amer. Chem. Soc., 1966, vol.88, p.4178
8. Mayor-Lopez M.J., Weber J., Mannfors B., Cunningham A.F. - Organomet., 1998, vol.17, p.4983
9. Несмеянов А.Н., Курсанов Д.Н., Сеткина В.Н., Кислякова Н.В. – Изв. АН СССР (Хим.Сер.), 1962, стр.1932
11. Woodward R.B., Rosenblum M., Whiting M.C. - J. Amer. Chem. Soc., 1952, vol.74, p.3458
12. Rozenblum M., Santer J.O. – J. Amer. Chem. Soc., 1959, vol.81, p.5517
13. Graham P.J., Lindsej R.V., Parshal J.V. – J. Amer. Chem. Soc., 1957, vol.79, p.3416
14. Broadhead G.D., Osgerby J.M., Pauson P.L. – J. Chem. Soc. 1958, p.650
15. Alper H., Kempuer S.H. – J. Org. Chem., 1974, vol.39, p.2303
16. Несмеянов А.Н., Пушин А.Н., Сазонова В.А. - Изв. АН СССР (Хим.Сер.), 1978, стр.1685
17. Aharoni S.M., Litt M.H. – J. Organometal. Chem., 1970, vol.22, p.171
18. Стукан Р.А., Юрьева Л.П. – ДАН СССР, 1966, т.167, стр.1311
19. Sanders J.R. – j. Chem Soc. Dalton Trans, 1975, p.2340
20. Prins R., Korswagen A.R., Kortbeek A.G.T.G. – J. Organometal. Chem., 1972, vol.39, p.335
21. Little W.F. – In: Scott A.F. Survey of progress in chemistry. Vol. I.N.Y.: L., 1963, p.147
22. Gubin S.P. – Pure and Appl. Chem., 1970, vol.23, p.463
23. Перевалова Э.Г.. Губин С.П., Смирнов С.А., Несмеянов А.Н. – Изв. ДАН СССР, 1964, т.155, стр.857
0 комментариев