Навигация
Металлический проводник (проводник I рода), соединяющий электроды и обеспечивающий прохождение тока между ними; он называется внешней цепью
79355
знаков
8
таблиц
5
изображений
3. Металлический проводник (проводник I рода), соединяющий электроды и обеспечивающий прохождение тока между ними; он называется внешней цепью.
Если электролит представляет собой токопроводящий раствор одного или нескольких веществ в воде или ином растворителе, то такие системы относятся к электрохимии водных или неводных растворов; если электролитом служит расплавленная соль (или смесь расплавленных солей и оксидов), эти системы относятся к электрохимии расплавов или расплавленных сред; если межэлектродное пространство заполнено газом — к электрохимии газов. Электрохимическая система может находиться в равновесном (рис. 2, а) или неравновесном (рис. 2, б, в) состоянии.
Электрохимическая система, производящая электрическую энергию за счет протекающих в ней химических превращений, называется химическим источником тока или гальваническим элементом (рис. 2, б). Здесь электрод, посылающий электроны во внешнюю цепь, называется отрицательным электродом или отрицательным полюсом элемента. Электрод, принимающий электроны из внешней цепи, называется положительным электродом или положительным полюсом элемента.
Электрохимическая система, в которой за счет внешней электрической энергии совершаются химические превращения, называется электролизером или электролитической ванной (рис. 2, в). Электрод, принимающий электроны от участников реакции, называется анодом. Электрод, отдающий электроны участникам реакции, — катодом. Часть электролита, примыкающая к аноду, называется анолитом; примыкающая к катоду — католитом.
Поскольку потеря электронов отвечает реакции окисления, а их приобретение — реакции восстановления, то можно сказать, что анод — это электрод, на котором происходит окисление, а катод — электрод, на котором происходит восстановление. Поэтому анод одновременно является отрицательным, а катод — положительным полюсом химического источника тока.
Изложенные соображения о различии электрохимических и химических реакций и о предмете и содержании электрохимии отвечают воззрениям, сложившимся в отечественной литературе. В согласии с расширенным определением электрохимии к ней можно отнести явления, связанные с электрохимическими свойствами коллоидов, с химическими реакциями, вызванными действием света или потока радиоактивных частиц (и приводящими к возникновению разности потенциалов), с электрохимическими явлениями в животных и растительных организмах и т. п. Представляется, однако, более правильным говорить в этих случаях о коллоидной электрохимии, фотоэлектрохимии, радиоэлектрохимии, биоэлектрохимии и т. д., сохранив название собственно электрохимии для
До сих пор рассматривалась роль, которую адсорбция играет лишь непосредственно в самом процессе электролитического восстановления (или окисления). Этот фактор должен сказываться на кинетике конкурирующих процессов, т. е. на кинетике электрохимического выделения водорода (или кислорода). Присутствие адсорбированных веществ на поверхности электрода может и увеличивать и уменьшать перенапряжение водорода (или кислорода). Это, в свою очередь, будет изменять условия протекания реакции восстановления (или окисления). Такого рода представления согласуются с тем экспериментальным фактом, что реакции электроокисления или электровосстановления многих веществ часто протекают со значительным выходом по току при потенциалах, заметно больших, чем те, при которых (при той же плотности тока) происходит выделение водорода или ирииелодит выделение водорода или кислорода из растворов, не содержащих «деполяризатора». В частности, проведение синтеза Кольбе возможно именно потому, что органические соединения, адсорбируясь на платиновом электроде, отравляют его, затрудняя тем самым выделение кислорода и смещая потенциал до значения, при котором уже может начинаться окисление анионов карбоновых кислот. Адсорбция органических веществ при столь положительных и удаленных от нулевой точки металла потенциалах кажется невозможной, если не учитывать окисления поверхности. Нулевая точка окисленного металла, как это было отмечено на примере свинца, сдвинута далеко в положительную сторону по сравнению с ее значением для чистого металла.
Глава 2. Электрохимическое выделение металлов 2.1. ОБЩАЯ ХАРАКТЕРИСТИКА ПРОЦЕССОВ
Электрохимическое выделение металлов из водных растворов и соединений лежит в основе гидроэлектрометаллургических процессов, т. е. процессов извлечения металлов из руд (электроэкстракцпя) и их очистки (рафинирование) при помощи электролиза. Гидро-электрометаллургическим путем получают и очищают такие металлы, как медь, никель, цинк, кадмий, олово, свинец, серебро, золото, марганец и др. Гидроэлектрометаллургия позволяет получать технически чистые металлы и в ряде случаев вести успешную переработку бедных руд. Электрохимическое выделение металлов использутся для защиты основного металла от разрушения при помощи покрытий из более устойчивых металлов или сплавов, а также для придания изделиям красивого, декоративного вида (гальванотехника). Кроме того, выделение металлов применяется для получения копий и воспроизведения художественных предметов, изготовления лент, бесшовных труб, печатных схем и т. д. (гальванопластика). Возможность использования процесса электролиза с выделением металлов для практических нужд была открыта в 1837—1838 гг. русским академиком Б. С. Якоби.
Электролитическое выделение металлов чаще всего проводят из растворов их простых солей — сульфатов, хлоридов или нитратов. Суммарной катодной реакцией в этом случае будет разряд гидра-тироваппых металлических ионов с их последующим переходом в кристаллическую решетку образующегося на катоде осадка:
ге-== [М] + * Н2О (22.1)
Электрохимическое выделение металлов из водных растворов происходит при более отрицательном потенциале, чем равновесный потенциал соответствующего металла в данных условиях. Разность между потенциалом электрода под током (при катодном выделении металла) и соответствующим обратимым электродным потенциалом дает электродную поляризацию
А #н = * J*-*!>• (22-2)
Долю общей поляризации, не связанную с замедленностью процессов транспортировки, часто называют перенапряжением металла. Перенапряжение и здесь тесно связано с природой электродного процесса.
Поляризация при электрохимическом выделении металлов, так же как и при других электродных реакциях, зависит от плотности тока, увеличиваясь вместе с ней. Однако в данном случае характер этой зависимости часто оказывается более сложным. Даже при осаждении одного и того же металла результаты поляризационных измерений могут укладываться в зависимости от диапазона применяемых плотностей тока, состава раствора и температуры на прямые в одной из следующих систем координат:
Ti — J. -n — lgJ, —~lg/. —r-lgj'
Экспериментальное исследование кинетики катодного выделения металлов представляет собой сложную задачу, что связано с некоторыми специфическими особенностями этого процесса. В ходе электролиза поверхность катода не постоянна, а непрерывно изменяется вследствие осаждения металла. Характер роста осадка существенно зависит от природы металла и условий электролиза
Для некоторых металлов, например серебра и таллия, типично образование нитеобразных кристаллов и древовидных ответвлений, так называемых усов и дендритов. При наблюдении за развитием отдельного нитеобразного кристалла можно обнаружить изменение его сечения, если меняется приложенный ток. Часто (рис. 22.1, а) с ростом силы тока нить утолщается, а при его уменьшении становится тоньше. Поверхность, на которой происходит осаждение, как бы приспосабливается к силе тока таким образом, чтобы плотность тока, а следовательно, и линейная скорость роста кристалла не менялись. Нередко наблюдается также слоистый рост осадка, при котором кристаллический пакет перемещается с определенной скоростью по поверхности катода (рис. 22.1, б). Металл осаждается в этом случае не на всей поверхности, а лишь на склоне пакета, который, таким образом, представляет собой действительный фронт роста кристалла. При исследовании условий образования осадка на монокристалле серебра было установлено, что устойчивый рост кристалла совершается по одной или несколько спиралям. На рис. 22.2 дана типичная микрокартина спирального роста осадка серебра
h _ h _ h _ h _ .
S\ S-2 S$ S4
и — распространение осадка по поверхности катода в виде толстого пакета

Рис. 22.1. Различные формы роста катодного осадка: а — изменение сечения растущей нити с изменением плотности тока
|
|

| Рис. 22.2. Микрофотография спирального роста осадка серебра |
Своеобразие роста электролитических осадков металлов затрудняет измерение плотности тока, иными словами, скорости электрохимического процесса. Здесь необходимо различать кажущуюся плотность тока, т. е. силу тока, приходящуюся на единицу геометрической (видимой) поверхности электрода, и истинную плотность тока, равную отношению силы тока к активной поверхности, т. е. к действительной поверхности роста осадка. В процессе образования катодного осадка при неизменной кажущейся плотности тока истинная плотность тока может меняться.
Изучение кинетики электроосаждения металлов связано также с затруднениями, возникающими в связи с неустойчивостью во времени потенциала катода. Изменение потенциала и электродной поляризации вызывается не только изменением активной поверхности и истинной плотности тока, по и другими причинами. Особенно заметно изменение потенциала со временем при выделении металлов на чужеродных электродах, когда электролиз приводит к образованию новой металлической фазы, например при осаждении кадмия, меди, серебра, ртути и ряда других металлов на платиновом катоде. Впервые это явление было обнаружено еще в 1910 г. Лебланом. Изменение величины перенапряжения со временем наблюдается при выделении металла и на одноименном катоде. На рис. 22.3 приведена типичная кривая поляризация -- время, полученная при выделении серебра на серебряном катоде.
По обычной методике снятия поляризационных кривых потенциалы измеряют через некоторый промежуток времени с момента наложения нового значения тока. В результате, как это следует из характера временного изменения потенциала (рис. 22.3), при одной и той же плотности тока получаются сильно отличающиеся значения поляризации, что затрудняет сопоставление данных, полученных разными авторами.
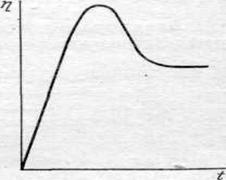
| Рис. 22.3. Изменение перенапряжения во времени, часто наблюдаемое при катодном выделении металлов |
Впервые это явление было обнаружено еще в 1910 г. Лебланом. Изменение величины перенапряжения со временем наблюдается при выделении металла и на одноименном катоде. На рис. 22.3 приведена типичная кривая поляризация - время, полученная при выделении серебра на серебряном катоде.
По обычной методике снятия поляризационных кривых потенциалы измеряют через некоторый промежуток времени с момента наложения нового значения тока. В результате, как это следует из характера временного изменения потенциала (рис. 22.3), при одной и той же плотности тока получаются сильно отличающиеся значения поляризации, что затрудняет сопоставление данных, полученных разными авторами.
Характер осадка и условия его формирования во времени при постоянной силе тока (или при заданном потенциале) зависят не только от природы металла, но и от состава раствора и присутствующих в нем примесей. Примеси поверхностно-активных веществ, а также различных окислителей (например, растворенного кислорода) влияют на кинетику электровыделения металлов. В зависимости от степени чистоты раствора и природы примесей могут меняться характер роста кристаллов, число центров кристаллизации, возникающих за единицу времени на единице поверхности катода, значение поляризации при данной плотности тока, характер ее изменения со временем и т. п. В тех случаях, когда катодный выход металла меньше единицы (электроотрицательные металлы, высокие плотности тока), возникают осложнения, связанные с изменением (обычно повышением) рН прикатодного слоя вследствие выделения водорода. Подщелачивание раствора вблизи катода благоприятствует процессам гидролиза солей металла с образованием его основных солей и гидроксидов, которые могут влиять на ход электроосаждения и включаться в катодный осадок.
2.2. ОБРАЗОВАНИЕ МОНОАТОМНЫХ СЛОЕВ МЕТАЛЛОВ ПРИ ПОТЕНЦИАЛАХ ПОЛОЖИТЕЛЬНЕЕ РАВНОВЕСНЫХК настоящему времени можно считать доказанным, что на подложке из металла Mi, отличного по своей природе от осаждаемого металла М2, в очень многих случаях процесс осаждения начинается с образования моноатомного слоя, а возникновение и развитие кристаллических зародышей совершается уже на этом слое. Осаждение первого монослоя металла на чужеродной подложке наблюдается при потенциале более положительном, чем равновесный потенциал выделяющегося металла в данном растворе, т. е. при данной активности его ионов. В связи с этим в зарубежной литературе широко используется термин «допотенциальное осаждение» (underpotential deposition), который при буквальном его переводе на русский не передает сущности явления, вместо него поэтому используются термины «осаждение при недонапряжении» или «дофазовое осаждение». По-видимому, первым, кто наблюдал эффект дофазового осаждения металлов, был Гевеши, работы которого относятся к 1912 г. Они были затем основательно забыты, и интерес к этой проблеме возродился лишь в 60—70-х годах, и сейчас она оказалась в центре внимания и электрохимиков и физиков. Очень интересные результаты были получены с применением циклической вольтамперометрии и тонкослойных электрохимических систем. На рис. 22.4 приведена циклическая /—£-кривая, описывающая осаждение таллия на серебряном электроде. Катодный пик тока наблюдается при потенциале примерно на 0,4 В положительнее равновесного потенциала таллия в растворе выбранного состава и отвечает образованию первого монослоя. Второй подъем тока приходится на равновесный потенциал таллия и соответствует выделению компактного осадка таллия, т. е. появлению в системе новой твердой фазы — металлического таллия. Переход в анодную область позволяет наблюдать максимум тока при равновесном потенциале таллия и другой максимум, отвечающий растворению дофазового слоя. Количество таллия, осевшего в области дофазового осаждения, можно с достаточной точностью рассчитать по количеству электричества, потраченного на его растворение при потенциале первого пика. Расчеты показывают, что при дофазовом осаждении образуются обычно полные или незавершенные моноатомные металлические слои. Так как потенциал дофазового осаждения (потенциал пика) ё>П положительнее соответствующего равновесного потенциала &р, то энергия связи атомов первого монослоя с чужеродной подложкой должна быть больше, чем атомов осаждаемого металла с одноименной подложкой. Сдвиг потенциала Дс?п в положительную сторону является следствием повышенной энергии связи A<?m,2 или, с некоторым приближением, повышенной теплоты адсорбции:
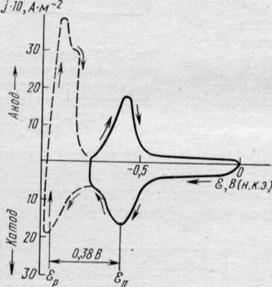
| Рис. 22.4. Циклическая вольтампер иая кривая, полученная на Ag-электроде в 2-Ю-4 М T1NO3 на фоне 0,5 М Na2SO,i; скорость развертки 20-Ю-3 В-с-1 |
Потенциал дофазового осаждения можно описать формулой Нерн-ста для электродов первого рода:
Если принять, что активность металла аи в растущем моноатомном слое отлична от единицы (как это обычно принимается для компактного металла) и зависит от степени покрытия в подложки монослоем,
Поскольку 02f2<l, потенциал монослоя в согласии с опытом оказывается положительнее равновесного потенциала того же металла в том же растворе. Из (22.5) также в согласии с опытом следует, что &П меняется с активностью ионов металла в растворе по такому же закону, как и равновесный потенциал электрода первого рода:
Соблюдение зависимости (22.6) указывает на то, что величина п отвечает заряду иона z, т. е. в образовании монослоя участвуют не ионы или частично разрядившиеся ионы, а атомы металла.
Уравнение (22.5), таким образом, удовлетворительно описывает ряд основных особенностей дофазового осаждения металлов, но не раскрывает его механизм. В этом отношении более перспективными представляются работы, в которых Д£„ сопоставляется с элект-роотрицательностями металлов, работами выхода электронов из них и т. д. По Кольбу, Прзасницкому и Геришеру, между Д£п и разностью работ выхода электрона a«'£ из металла подложки (Mi) и из осаждающегося металла (М2) существует прямо пропорциональная зависимость с наклоном, равным 0,5:
F\g]{= 0,5 Ды1'! . (22.7)
Подобная же зависимость была найдена Трасатти, ко с наклоном, равным единице:
F \ё „ = Wgl . (22.8)
Рассмотрение кривых Д£ —Да>£- с привлечением всех наиболее надежных данных по потенциалам дофазового осаждения и работам выхода показало, что коэффициент пропорциональности k лежит между 0,5 и 1, т. е.
F \(о и — k Дсо ' . /99 Q\
Независимо от величины k из уравнений (22.7) —(22.9) следует, что дофазовое осаждение металлов наблюдается только в том случае, когда работа выхода электрона из металла подложки (металл Mi) больше, чем из металла монослоя (М2). Следовательно, образование монослоя сопровождается переносом электронов из него в субстрат и появлением диполей на границе раздела М, и М2, причем положительный конец диполя расположен на монослое. Свойства монослоя, его структура, во многом определяемая структурой субстрата, играют очень важную роль в процессе дальнейшего развития осадка, влияя также на адсорбционные, каталитические, коррозионные и другие характеристики металла. Дофазовое осаждение представляет поэтому не меньший интерес, чем. зароды-шеобразование, и с ним необходимо считаться при рассмотрении механизма возникновения новой металлической фазы.
Несмотря на все особенности протекания процессов электрохимического выделения металлов, создающие серьезные трудности при проведении экспериментов и при теоретической интерпретации их результатов, к настоящему времени уже накоплен значительный фактический материал и сформировались определенные взгляды на природу этих процессов. Получение достаточно достоверных опытных данных сделалось возможным благодаря развитию техники эксперимента (применение новых методов исследования, при помощи которых удается избежать осложнений, связанных с особенностями построения кристаллической решетки и изменением
потенциала во времени), разработке методики измерения поверхностен роста и, соответственно, истинной плотности тока, тщательной очистке растворов и т. п.
2.3. ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ПРОЦЕССЫ КАТОДНОГО ВЫДЕЛЕНИЯ МЕТАЛЛОВ 2.3.1. Роль природы металлаПри электролизе растворов простых солей характер катодных осадков и величина электродной поляризации определяются в первую очередь природой выделяющегося металла (табл. 22.1).
Таблица 22.1. Классификация металлов по значению металлического перенапряжения при их выделении из растворов простых солей
| группы | Металлы | Перенапряжение, В | 2 Ток обмена, А-м | Средние размеры зерен осадка, м |
| I | Hg, Ag, Tl, Pb, Cd, Sn | 0—П-1Э-3 | п-103- п-13 | >10-5 |
| II | Bi, Cu, Zn | п -13-2 | /;. 13—1!. 13-1 | 10-5—Ю-6 |
| III | Co, Fe, Ni | п-13-1 | п-13-4—п-13-5 | <10-7 |
Все металлы, приведенные в табл. 22.1, можно разделить на три группы. К первой из них относятся металлы, выделяющиеся из водных растворов или совсем без перенапряжения (ртуть), или с очень малым перенапряжением, не превышающим при обычных плотностях тока тысячных долей вольта (серебро, таллий, свинец, кадмий, олово). Для этой группы металлов (кроме ртути) наиболее отчетливо проявляются неустойчивость потенциала во времени, сложный характер роста катодного осадка и другие особенности, свойственные процессу катодного выделения металлов. При промышленных плотностях тока эти металлы дают грубые осадки. Токи обмена для металлов этой группы очень велики. Так, например, ток обмена между металлической ртутью и раствором ее нитрата превышает 103 А-м-2, а между серебром и раствором нитрата серебра достигает 102 А-м~2.
Висмут, медь, цинк образуют вторую, промежуточную группу. Для нее характерно металлическое перенапряжение порядка нескольких десятков милливольт, образование более тонких осадков и меньшие, чем у металлов предыдущей группы, токи обмена. Для меди, например, ток обмена в контакте с раствором сульфата меди близок к Ю-1 А-м-2.
Наибольшим металлическим перенапряжением обладают металлы третьей группы, у которых оно достигает нескольких десятых долей вольта. Эти металлы выделяются на катоде в виде плотных тонкокристаллических осадков. Токи обмена у них малы, составляя для железа и никеля в растворах их сульфатов соответственно 10-4и 10-5А-м-2.
Данные, приведенные в табл. 22.1, относятся к обычным условиям электролиза, когда металл выделяется на поликристаллической основе и дает отложения, также имеющие поликристаллическую структуру. Поверхность таких осадков образована гранями с различными кристаллографическими индексами. В зависимости от режима электроосаждения на поверхности осадка могут преобладать те или иные грани. Поэтому важно выяснить, зависит ли металлическое перенапряжение от того, на какой грани выделяется металл. Опыты с монокристаллами ряда металлов, ориентированными по отношению к раствору различными граниями, подтвердили существование подобной зависимости (см. табл. 22.2).
Таблица 22.2. Зависимость металлического перенапряжения (В• 103) от природы грани монокристалла (при 10 Ам~2, / = 25° С)
| Индекс грани | ||||
| Металл | Раствор | |||
| (100) | (ПО) | (Ш) | ||
| РЬ | 0,5 М РЬ(СЮ4)2, | 3,0 | 3,0 | 4,4 |
| 0,5 М НС1О4 | ||||
| Sn | 0,5 М SnCl2 | 2,5 | 4,0 | _____ |
| 0,5 М НС1 | ||||
| Си | 0,5 М Си(С1О4)>, | 35 | 40 | 43 |
| 0,5 М НС1О4 | ||||
| Ni | 10 М NiCl2, | 768 | 839 | 819 |
| 0,33 М Н3ВО3 | ||||
| (рН 3,1) | ||||
Из табл. 22.1 и 22.2 следует также, что значение металлического перенапряжения в большей степени определяется природой металла, чем кристаллографической ориентацией электродной поверхности. Независимо от того, на какой из граней происходит выделение металла, перенапряжение всегда выше для никеля, чем для меди, а для меди оно всегда больше, чем для олова или свинца.
На подчиненную роль кристаллизационных факторов в явлениях перенапряжения указывают также данные по кинетике катодного выделения растворимых в ртути металлов на соответствующих амальгамах. Результаты кинетического исследования реакций обмена металлическими ионами между разбавленными амальгамами и растворами нитратов указывают на уменьшение тока обмена в следующем ряду:
TI, Pb, Cd>Cu, Zn>Ni
Характер кривых потенциал — время, полученных Гейровским методом осциллографической полярографии, показывает, что степень обратимости реакции разряда и ионизации на ртутных (точнее амальгамных) электродах уменьшается в последовательности TI, Pd, Cd, Sn, Bi, Sb, Sb, Cu
Порядок расположения металлов по степени их необратимости, а следовательно, по величине металлического перенапряжения практически не зависит от того, осаждается ли металл на твердом одноименном катоде или на разбавленной амальгаме соответствующего металла. Выделение металлов группы железа и на ртутном катоде сопровождается значительно большей поляризацией, чем у всех других металлов, приведенных в табл. 22.1. Оно протекает здесь еще менее обратимо, чем на твердых катодах. Однако эти металлы почти не способны образовывать амальгамы, и их осаждение в случае применения ртутных катодов совершается на плохо связанных между собой мелких кристаллических островках.
2.3.2. Роль состава раствораСистематические исследования влияния состава раствора на кинетику электроосаждения металлов были начаты в 1917 г. Н. А. Из-гарышевым. Было установлено, что при катодном выделении металлов из растворов их простых солей существенное значение имеет природа аниона соли. Влияние природы аниона на перенапряжение и на характер образующихся осадков наблюдается для многих металлов, но наиболее сильно оно проявляется для металлов, выделение которых не сопровождается высокой поляризацией. Обычно перенапряжение уменьшается при переходе от одного аниона к другому в следующем порядке:
POJj-, №^, SO2-, C104->NH2S03~>Cl->Br->I-
Причем в том же направлении увеличивается тенденция к образованию более грубых, крупнокристаллических осадков. Влияние анионов вполне сравнимо с эффектами, связанными с кристаллографическими факторами. Так, например, замена перхлоратных растворов на сульфаминовые уменьшает перенапряжение при выделении свинца примерно в той же степени, как и переход от грани (111) к грани (ПО).
Присуствие в растворе, помимо ионов разряжающегося металла, «индифферентных» катионов увеличивает металлическое перенапряжение. Подобные эффекты наблюдались при выделении никеля, цинка, меди и других металлов. В водных растворах обычными «посторонними» катионами являются водородные ионы. Увеличение их концентрации приводит чаще всего к росту металлического перенапряжения. Значительное его повышение наблюдается в присутствии поверхностно-активных катионов типа тетразамещенного аммония.
Высокая чувствительность процесса электроосаждения металлов к чистоте растворов указывает на то, что присутствие не только электролитов, но и любых веществ, особенно обладающих поверхностно-активными свойствами, должно играть здесь существенную роль. Так, введение в ванну цинкования ничтожного количества желатины (порядка 0,005%) изменяет величину катодный поляризации и характер получающихся осадков (Н. А. Йзгарышев, П. С. Титов, 1917).
Введение в раствор небольших количеств молекулярных и ионных веществ — один из наиболее эффективных способов воздействия на ход процесса электроосаждения металлов. Многие, преимущественно органические, вещества способны увеличивать блеск осадков (блескообразователи), сглаживать их поверхность (выравниватели), и изменять другие свойства, например пористость, твердость, хрупкость, способность окклюдировать водород и т. д. (Кудрявцев, Матулис и др.).
Обнаруженная М. А. Лошкаревым адсорбционная поляризация проявляется в том, что при добавлении к раствору некоторых поверхностно-активных веществ (например, трибензиламина) изменяется скорость выделения металла на ртутном и на твердых катодах. Она становится, во-первых, меньше той, что наблюдалась до введения добавки, и, во-вторых, не зависящей в широкой области потенциалов от катодного потенциала. Однако после того как достигается определенный (обычно весьма отрицательный) потенциал, действие добавки прекращается. Скорость выделения начинает быстро расти, приближаясь к нормальному для этих условий значению, отвечающему предельному диффузионному току. Сопоставление результатов поляризационных измерений на ртутных катодах с электрокапиллярными кривыми и кривыми дифференциальной емкости (снятыми до и после введения добавки) показали, что потенциал, при котором прекращается действие добавки, совпадает с потенциалом ее десорбции (рис. 22.5). Действие добавки оказывается при этом специфическим. Одни и те же добавки или определенная их комбинация в разной степени тормозят разряд различных ионов на ртутном катоде. Явление адсорбционной поляризации используется для улучшения качества гальванических осадков при электролитическом получении сплавов.
Все эти данные относятся к тому случаю, когда металлы выделяются из растворов их простых солей. Если неорганические или органические добавки образуют комплексные соединения с выделяющимся металлом, то ход катодного процесса существенно меняется. Прежде всего образование комплексов в растворе смещает равновесный потенциал металла в отрицательную сторону за счет уменьшения концентрации его свободных ионов. Добавление вещества М. А (анионы которого способны давать комплексные соединения МАХ с ионами выделяемого металла Мг+) вызывает в растворе реакцию комплексообразования:
Мг+ +х А- = МА£"* , (22 10)
где (г—х)—заряд образующихся ионов. Реакции (22.10) отвечает константа комплексообразования
Обратная ей величина называется константой нестойкости комплекса
Константа нестойкости характеризует способность комплекса с диссоциации с регенерацией исходных ионов Mz+ и, таким образом, определяет их равновесную концентрацию.
В результате реакции комплексообразования определенная доля (юнов Мг+ (тем большая, чем ниже константа нестойкости) будет присутствовать в растворе в виде сложных ионов MA*""* и, следовательно, концентрация свободных ионов металла должна уменьшиться. Это уменьшение и, соответственно, сдвиг эбратимого потенциала электрода в отрицательную сторону будут тем значительнее, чем меньше констан-га нестойкости и чем выше концентрация добавки. Подбирая соответствующие комплексообразо-вателн и их концентрации, можно изменить равновесные потенциалы присутствующих в растворе попов различных металлов таким образом, чтобы обеспечить или их совместное осаждение в виде сплава, или наиболее полное разделение.
Появление комплексов в растворе сказывается не только на равновесных потенциалах металлов, но и на величине перенапряжения и на характере катодных осадков. При переходе от простых электролитов к комплексным обычно наблюдается повышение перенапряжения и уменьшение зернистости осадков; одновременно подавляется тенденция к образованию и росту дендритов. Так, серебро, которое при электролизе раствора его нитрата выделяется на катоде почти без поляризации и дает грубые, шероховатые осадки, из комплексных цианистых электролитов может быть получено в виде гладких тонкокристаллических отложений.
2.4. ПРИРОДА МЕТАЛЛИЧЕСКОГО ПЕРЕНАПРЯЖЕНИЯЭлектродная поляризация, наблюдаемая при выделении металлов, может быть связана либо с фазовыми превращениями (см. гл. 16) и представлять собой один из видов фазового перенапряжения (за-медленость образования трехмерных и двухмерных зародышей, поверхностная диффузия адатомов или аднонов), либо с замедленностью собственно электрохимической стадии (см. гл. 17) и совпадать с электрохимическим перенапряжением. При осаждении металлов существенную роль играют затруднения на стадии транспортировки, а также на стадии химического превращения (см. гл. 15), предшествующего электрохимическому акту. При рассмотрении процессов катодного выделения металлов (особенно из комплексных электролитов) необходимо поэтому всегда учитывать концентрационную поляризацию, т. е. диффузионное перенапряжение и химическое или реакционное перенапряжение. Наконец, в условиях катодного выделения металлов энергетическое состояние иона в образующемся осадке может отличаться от его состояния в нормальной кристаллической решетке данного металла и, как правило, отвечать более высокому уровню энергии. Переход из такого мета-стабильного состояния к обычному также может обусловливать появление особого вида фазового (кристаллизационного) перенапряжения.
Преобладание того или иного вида перенапряжения определяется природой металла, составом раствора, плотностью тока, температурой электролита. При обычных температурах и при использовании простых, некомплексных электролитов перенапряжение изменяется с природой металлов, как это показано в табл. 22.1. Опытные данные указывают на то, что выделение металлов, стоящих в начале ряда (Hg, Ag, Tl, Pb, Cd, Sn), сопровождается лишь незначительной поляризацией, связанной главным образом с замедленностью возникновения и развития новой фазы. Замедленность электрохимической стадии не играет здесь существенной роли. В электрохимической литературе эти металлы, для которых характерно фазовое перенапряжение, называются часто нормальными металлами. Напротив, при выделении металлов, стоящих в конце ряда табл. 22.1 (металлы группы железа), наблюдается высокая поляризация; обусловленная преимущественно замедленностью электрохимической стадии. Эти металлы, для которых характерно электрохимическое перенапряжение, называются инертными металлами. Промежуточное положение и по величине поляризации, и по природе перенапряжения (здесь наиболее вероятно наложение нескольких видов перенапряжения) занимают такие металлы, как Bi, Си и Zn.
Это различие в величине и механизме перенапряжения обусловливает, согласно Фольмеру, различный характер осадков, в виде которых нормальные и инертные металлы выделяются па катоде. Все факторы, вызывающие торможение акта разряда, должны, с этой точки зрения, уменьшать относительную роль кристаллизационных явлений и приводить к получению равномерных мелкозернистых осадков. Увеличение торможения достигается или переводом простых ионов в более прочные комплексы, или при помощи добавок поверхностно-активных веществ (если их адсорбция больше всего сказывается на акте разряда). Изменение структуры осадков, наблюдаемое при переходе от простых электролитов к цианистым, а также характер электроосаждения в условиях адсорбционной поляризации подтверждают эту точку зрения.
2.5. ФАКТОРЫ, ОПРЕДЕЛЯЮЩИЕ ВЕЛИЧИНУ ПОЛЯРИЗАЦИИ ПРИ КАТОДНОМ ВЫДЕЛЕНИИ РАЗЛИЧНЫХ МЕТАЛЛОВ 2.5.1. Энергия иона в металле и его состояние в раствореДо сих пор остается недостаточно ясным, почему существует такое большое различие в величине и природе металлического перенапряжения для нормальных и инертных металлов и с какими свойствами металлов (или растворов) оно связано. Была сделана попытка объяснить эти явления различным соотношением между прочностью связи ионов в растворе и в кристаллической решетке нормальных и инертных металлов. Подобное предположение эквивалентно допущению, что в разряде участвуют ионы в той форме, в какой они присутствуют в растворе, и что разряд переводит ион непосредственно в его конечное положение в решетке металла.
При таком допущении энергия активации должна быть функцией энер'гии гидратации ионов и работы их выхода из металла, возрастая с увеличением разности между ними. На основании этого следовало бы ожидать, что для инертных металлов энергия гидратации больше, а работа выхода меньше, чем для нормальных металлов. Однако имеющиеся данные (см. табл. 22.3) не подтверждают такого предположения: так, для цинка и никеля значения энергии гидратации и работы выхода почти одинаковы, но цинк выделяется со значительно меньшим перенапряжением, чем никель. Это отнюдь не означает, что прочность ионов в растворе и в металле не играет никакой роли, ее просто нельзя учесть подобным примитивным способом.
Недостаточность такой упрощенной картины следует и из более общих соображений. Стандартные электродные потенциалы изменяются параллельно с изменением разности между химической' энергией гидратации ионов в растворе и работой удаления ионов из кристаллической решетки. Поэтому казалось бы, что металлическое перенапряжение должно изменяться в такой же последовательности, как и ряд стандартных электродных потенциалов. Опыт противоречит этому выводу. Так, например, цинк, стандартный потенциал которого равен —0,76 В, выделяется с меньшим перенапряжением, чем железо со стандартным потенциалом —0,44 В. В то же время перенапряжение при выделении цинка примерно такое же, как и при выделении меди — электроположительного металла (<r ^U2+ (Cu = + 0,34В).
Таблица 22.3. Энергия гидратации и работы выхода ионов для ряда металлов (в кДжмоль"1)
| Свойства | Нормальные | и переходные металлы | Инертные металлы | |||||
| Ag + | Cd2+ | Pb2+ | CU2 + | Zn2+ | Со2+ | Fe2+ | N,2+ | |
| v+ | 477 498 | 18)4 18)8 | 1498 1553 | 2082 2240 | 2)3) 1926 | 2024 1712 | 1920 180) | 2)74 1976 |
Допущение о том, что выделение металла совершается не как последовательная стадийная реакция, а как один элементарный акт, противоречит всем результатам, полученным при изучении кинетики различных электрохимических процессов. Например, для реакции катодного выделения водорода принятие такого допущения привело бы к не отвечающему действительности выводу о независимости водородного перенапряжения от природы металла. Чтобы объяснить связь, существующую между металлическим перенапряжением и природой металла, а также характер влияния состава раствора на величину перенапряжения, необходимо принимать во внимание не только начальное и конечное состояния металлических ионов, но и природу элементарных актов. При этом следует учитывать состояние и свойства реагирующих частиц на разных стадиях суммарного процесса.
Многие исследователи пытались усовершенствовать теорию электровыделения металлов, привлекая представления об электронном строении их ионов. Одна из таких попыток принадлежит Лайон-су (1954). По Лайонсу, величина металлического перенапряжения зависит от характера электронных структур разряжающихся ионов и выделившегося на катоде металла. При этом перенапряжение будет особенно большим в двух случаях. Во-первых, если аквакомп-лексы (или иные комплексы) образованы ионами за счет электронов, находящихся на внутренних орбитах (внутриорбитальные комплексы), благодаря чему создаются наиболее прочные связи ионов в растворе. Во-вторых, если велика разница в электронных структурах иона и металла; в этом случае требуется значительная энергия активации для их перестройки в процессе разряда. Разряжающиеся ионы имеют обычно иную структуру, чем ионы, присутствующие в растворе. Это обстоятельство связано с тем, что при адсорбции на электроде происходит частичная диссоциация простого гидратированного или комплексного иона. Освобождающиеся связи удерживают ион на поверхности электрода, причем до момента разряда идет дальнейшая перестройка его электронной структуры в направлении, сближающем ее со структурой иона в металле. Далее следует разряд или с полной дегидратацией иона, или с распадом комплекса и внедрением атома металла в кристаллическую решетку.
Все металлы, приведенные в табл. 22.1, дают, по Лайонсу, впеш-неорбитальные аквакомплексы, т. е. комплексы, образованные с участием электронов, находящихся на внешних орбитах. Это делает возможным их выделение из водных растворов в отличие от таких металлов, как титан, цирконий и др., ионы которых в растворе присутствуют в виде внутриорбитальных комплексов. Высокое перенапряжение металлов группы железа объясняется тем, что электронная структура их разряжающихся акваионов значительно отличается от структуры соответствующего металла. Электронные структуры нормальных металлов в кристалле и в водных комплексах близки, и поэтому они обладают низким перенапряжением.
Взгляды Лайонса в какой-то мере отражают некоторые особенности, свойственные катодному выделению металлов. Несомненно, что известная роль в этих процессах должна быть отведена специфике электронного строения ионов. Однако этого недостаточно для полного выяснения природы процессов электроосаждения металлов. Прежде всего это связано с отсустствием надежных данных о строении ионов в растворе и на поверхности электрода, что заставляет прибегать к помощи гипотетических структур. Далее, теория Лайонса даже при использовании подобных структур не в состоянии объяснить некоторые полученные экспериментально закономерности, относящиеся, например, к выделению металлов группы платины. В его теории не учитывается влияние потенциала электрода и строения двойного электрического слоя на процесс электроосаждения металла. Наконец, она не может объяснить той роли, какую играют в этом процессе состав раствора и особенно поверхностно-активные вещества. Дальнейшее развитие представлений о роли структуры разряжающихся металлических ионов при электроосаждении металлов было дано Вылчеком (1957).
2.5.2. Активность поверхности катода в процессе осаждения металлов
Согласно другой точке зрения природа и величина металлического перенапряжения являются функцией состояния поверхности катода, которое может быть неодинаковым для разных металлов. Одна из причин этого различия связана с возможностью выделения
водорода и его влиянием на ход осаждения металлов. Известно, что электролитические осадки железа, никеля и кобальта всегда содержат заметное количество водорода. Включения водорода можно рассматривать как одну из возможных причин искажения кристаллической решетки осадков этих металлов, появления в них внутренних натяжений, хрупкости и т. п. В меньших количествах водород присутствует в осадках меди и цинка. Его практически не удается обнаружить в электролитически осажденных кадмии или свинце. Из этого следует, что металлическое перенапряжение увеличивается параллельно с количеством водорода, включенного в осадок металла, т. е. водород, по-видимому, затрудняет процесс катодного выделения металла. Предполагалось, что водород выступает здесь в роли отрицательного катализатора, тормозя разряд за счет создания поверхностной пленки или образования гидридов металлов.
В связи с влиянием водорода на кинетику электроосаждения металлов важно выяснить причины, которые приводят к различному содержанию водорода в разных металлах и, следовательно, изменяют величину его тормозящего действия при переходе от одного металла к другому. Оказалось, что в общем случае нет прямой зависимости между долей общего тока, расходуемой на выделение водорода, и его содержанием в металле. Так, например, при электроосаждении цинка выход по току водорода обычно больше, чем в случае железа; тем не менее содержание водорода в нем всегда меньше и перенапряжение при его выделении ниже. Расположение металлов в порядке увеличения перенапряжения при их выделении примерно соответствует их расположению по степени уменьшения водородного перенапряжения. Однако большее значение должна иметь не величина перенапряжения водорода, а механизм его выделения на данном металле (Л. И. Антропов, 1952). Включение водорода в осадок металла тем вероятнее, чем медленнее протекает удаление адсорбированных водородных атомов с поверхности металла. Наибольшие количества водорода обнаруживаются поэтому в катодных осадках металлов группы железа, где стадия рекомбинации водородных атомов протекает медленно.
Присутствие адсорбированного или окклюдированного водорода все-таки не может считаться главным фактором, определяющим специфику катодного выделения металлов.
Причину различия в значениях металлического перенапряжения и в характере катодных осадков можно было бы искать в неодинаковой склонности металлов к пассивированию и в их разной адсорбционной способности. Появлениен на поверхности растущего осадка посторонних.веществ затрудняет и разряд металлических ионов, и их внедрение в кристаллическую решетку. Этот тормозящий эффект должен быть тем заметнее, чем легче пассивируется данный металл. Пассивирующими агентами могут быть растворенный кислород, примеси органических соединений и каталитических ядов, некоторые посторонние ионы, не участвующие непосредственно в электродной реакции, и другие вещества. Особое положение металлов группы железа, в частности их высокое металлическое перенапряжение, объясняется с этой точки зрения тем, что они в большей мере, чем другие металлы, склонны к пассивированию. Однако и этот фактор не является, по-видимому, решающим и не обусловливает порядка расположения металлов по величине их перенапряжения. Даже после самой тщательной очистки растворов от примесей и удаления из них кислорода разница в значениях металлического перенапряжения между инертными и нормальными металлами остается большой. Точно так же свинец, который пассивируется несравненно легче, чем цинк, выделяется при более низком перенапряжении.
2.5.3. Заряд поверхности металла в условиях его катодного осаждения
Протекание адсорбционных явлений на границе металл — электролит, а следовательно, В степень их влияния на процессы, идущие на этой границе, во многом зависит от потенциала, или точнее, от заряда металла.
Ранее считалось, как само собой разумеющееся, что поверхность катода всегда отрицательна, причем тем более отрицательна, чем менее электроположителен электродный металл. Эта точка зрения, сохранившая известное распространение и в настоящее время, ошибочна. Заряд поверхности металла не определяется ни той ролью, какую металл играет в электрохимическом процессе (т. е. является ли он катодом или анодом), ни его электродным потенциалом в данных условиях. Заряд поверхности электрода можно оценить, если воспользоваться предложенной Л. И. Антроповым приведенной, или ср-шкалой потенциалов. Потенциал электрода в ф-шкале представляет собой разность между его потенциалом в данных конкретных условиях (например, в процессе электроосаждения металла) и соответствующей нулевой точкой. Потенциал электрода в приведенной шкале служит мерой заряда поверхности и позволяет предвидеть, адсорбция каких именно ионов будет наиболее вероятной в данных условиях. Это положение можно проиллюстрировать на примере катодного выделения никеля, цинка, кадмия и свинца из растворов их простых солей. Все эти металлы выделяются при отрицательных потенциалах (по водородной шкале), которые в обычных режимах электролиза имеют следующие значения: —0,80 В (№), —0,80 В (Zn), —0,45 В (Cd) и —0,15 В (РЬ). Их потенциалы в приведенной шкале, т. е. заряды, можно оценить, воспользовавшись данными о нулевых точках этих металлов (см. табл. 11.6):
?М, ==-0,83 —( —0,2) = -0,6В;
<PZn = - 0,80 — (- 0,5) = - 0,3 В;
?Cd = - 0,45- ( - 0,7) = + 0,25 В;
9РЬ = — 0,15 — (-0,7) ==+0,55 В.
Из этих значений ф-потенциалов следует, что в условиях электроосаждения никель имеет наиболее отрицательный заряд, за ним следует цинк, который также заряжен отрицательно, в то время как кадмий и свинец заряжены положительно. В ходе электровыделения на поверхности осадков никеля и цинка должны , адсорбироваться преимущественно катионы, а на поверхности осадков кадмия и свинца — главным образом анионы. Эти выводы, строго говоря, справедливы течением концентрации водородных ионов и при введении поверхностно-активных катионов, резкое увеличение перенапряжения водорода при переходе от положительно заряженной поверхности металла к отрицательно заряженной и т. д. Все нормальные металлы (Hg, Ag, Tl, Pb, Cd), при выделении которых перенапряжение ничтожно мало, заряжены положительно по отношению к растворам их простых солей (ф>0), а все металлы, выделение которых сопровождается высоким перенапряжением (металлы группы железа), — отрицательно (ф<0). Поэтому в явлениях электроосаждения металлов необходимо учитывать заряд поверхности электрода, хотя он и не определяет всех особенностей этих процессов. Так, например, неясным остается факт существования большой разницы в значениях перенапряжения при выделении цинка и никеля — металлов, обладающих в условиях равновесия приблизительно одинаковым отрицательным зарядом поверхности. Точно так же выделение меди, судя по ее ф-потенциалу, должно бы происходить с такой же легкостью, как и выделение кадмия или свинца, тогда как данные опыта противоречат этому.
2.5.4. Другие возможные причины появления металлического перенапряжения
Невозможность объяснить все кинетические особенности электрохимического выделения металлов с какой-либо одной общей точки зрения заставляет искать новые пути истолкования этих процессов и прибегать к предположениям частного характера. Так, например, существует мнение, что перенапряжение при выделении металлов связано с числом электронов, участвующих в элементарном акте разряда (Гейровский). При этом предполагают, что одно электронные реакции протекают практически без торможения.
В тех случаях, когда только один электрон участвует в акте разряда (или когда процесс можно разбить на ряд последовательных одноэлектронных стадий), перенапряжение должно быть низким. Если в разряде ионов металла участвуют одновременно два электрона, то следует ожидать появления высокого металлического перенапряжения. Согласно этим представлениям низкое перенапряжение, наблюдаемое при выделении таллия и серебра, связано с тем, что реакция восстановления требует участия одного электрона:
Ag+ + е- = [Ag]
Невысокое металлическое перенапряжение, свойственное меди и цинку, объясняют возможностью протекания разряда в две одно электродные стадии:
CU2+ +е~ = Си+ Cu+ + e~ = [Си]
е~ = Zn+
Zn+ ■+• е~ = [Zn]
Для металлов группы железа разряд совершается при одновременном присоединении двух электронов чем обусловливаются малая скорость этого процесса (вероятность одновременного присоединения двух электронов низка) и высокое перенапряжение.
2е~ = [Fe]
Однако уже давно было замечено, что скорость электроосаждения, а также электрорастворения металлов группы железа зависит от рН раствора и присутствия в нем примесей. Р. X. Бурштейн, Б. Н. Кабанов и А. Н. Фрумкин (1947) высказали предположение о непосредственном участии ионов ОН~ в кинетике этих процессов. По их мнению, ионы ОН~ играют роль своеобразных катализаторов. Механизм реакций катодного осаждения и анодного растворения железа, кобальта и никеля с образованием промежуточных частиц типа FeOH, FeOH+ или Fe-FeOH+ рассматривался затем Хейслером, Бокрисом, Фишером и Лоренцом и многими другими авторами. Было предложено несколько схем, объясняющих такие экспериментальные данные, как характер зависимости скорости реакции от рН, небольшой наклон тафслсвской прямой в чистых растворах серной кислоты, его повышение при переходе к растворам соляной кислоты и при введении добавок поверхностно-активных веществ и т. д. В качестве иллюстрации можно привести схему Бокриса
Fe2+ + ОН- = FeOH+ FeOH+ + e~ = FeOH
— [Fe] + O
СМ ворд док – глюк
Заключение
Лаборатория электрокатализа и коррозии (зав. лабораторией — д.х.н., профессор О.А.Петрий)В лаборатории изучается широкий круг электрокаталитических и коррозионных явлений на металлах, сплавах, оксидах и различных композиционных материалах в водных и апротонных средах. В основе подхода к этим исследованиям лежит экспериментальное и теоретическое рассмотрение процессов на микроскопическом уровне, поэтому наряду с электрохимическими методами используются и совершенствуются применительно к новым объектам и задачам методы сканирующей туннельной микроскопии и спектроскопии, эллипсометрии, а также комплекс физико-химических методов структурного анализа материалов. Разрабатываются нетрадиционные способы получения наноструктур и многофазных композиций, основанные на процессах анодной и катодной электрокристаллизации. Развиваются новые подходы к молекулярному дизайну поверхностей электродов-катализаторов.
В теоретическом плане важнейшими задачами лаборатории являются развитие теории электрокатализа, представлений о механизмах электрокаталитических процессов, а также установление закономерностей элементарного акта переноса электрона в гетерогенных системах, в первую очередь экспериментальная проверка квантово-механической теории элементарного акта с использованием нетрадиционных модельных реакций. Развиваются представления о влиянии заряда электрода на различные кинетические параметры и рассматриваются на микроскопическом уровне эффекты двойного электрического слоя в электрохимической кинетике. На основе экспериментальных исследований электродов с обновляемой поверхностью и компьютерного моделирования развиваются представления о реконструкции многокомпонентных поверхностей и о процессах поверхностной диффузии.
С целью разработки никель-металлогидридных аккумуляторов проводится широкий поиск материалов АВ5- и АВ2-типов, обратимо сорбирующих водород в электрохимических условиях.
С 1997 г. лаборатория входит вместе с Институтом физической химии РАН и кафедрами физической и коллоидной химии в состав Учебно-научного центра «Нанохимия».
Последние публикации
1. O.A.Petrii, G.A.Tsirlina, Electrochemistry of Oxide High-Temperature Superconductors, in «Adv. Electrochem. Sci. Eng.»/Ed. R.C.Alkire, H.Gerisher, D.M.Kolb and C.W.Tobias, 1997, V.5, P.61-123.
2. R.R.Nazmutdinov, G.A.Tsirlina, Yu.I.Kharkats, O.A.Petrii, M.Probst, Activation Energy of Electron Transfer between a Metal Electrode and Reagents of Nonspherical Form and Complicated Charge Distribution. Cr(EDTA) Complexes//J.Phys. Chem, 1998, V.102, P. ??-??.
3. V.A.Safonov, M.A.Choba, Yu.D.Seropegin, Peculiarities of the Electrical Double Layer Structure on Renewed Electrodes of Eutectic Alloys//Electrochim. Acta, 1997, V.42, P.1907–1914.
Литература:
Основная:
1. Дамаскин Б.Б., ПетрийО.А., Основы теоретической электрохимии,
М., Высшая школа, 1978
2. Дамаскин Б.Б. , О.А. Петрий Введение в электрохимическую кинетику, 1986 г.
3. Антропов Л.И., Теоретическая электрохимия , М.,Высшая школа, 1975 г.
4. Скорчелетти В.В., Теоретическая электрохимия ,М.. Высшая школа, 1974г.
5. Ротинян А.Л., Тихонов К.И. ,Шонина И.А. Теоретическая электрохимия, Высшая школа 1980 г.
6. Попова С.С. Анодное растворение металлов и пассивация в кислых окислительных средах.- Саратов ,Изд-во СГУ, 1984 г.
7. Кукоз Ф.И. Сборник задач по теоретической электрохимии.
8. Попова С.С. Теоретическая электрохимия. Сборник задач.
9. Левин А.И., Помосов А.В. Лабораторный практикум по теоретической электрохимии.- М: Металлургия, 1979 г.
10. Дамаскин Б.Б.Практикум по электрохимии-М: Высшая школа, 1991г.
Дополнительная
1. Феттер К., Электрохимическая кинетика, М.,Л., «Химия», 1967г.
2. Фрумкин А.Н., БагоцкийВ. С., ИофаЗ.А., Кабанов Б.Н., Кинетика электродных процессов , изд-во МГУ, 1952г.
3. Дамаскин Б.Б., Петрий О.А. , Батраков В.В., Адсорбция органических соединений на электродах. М.-Л., «Наука», 1968г.
4. Дамаскин Б.Б., Принципы современных методов изучения электорохимических реакций, Изд-во МГУ ,1965г.
5. Делахей П., Новые приборы и методы в электрохимии .,ИЛ, 1957г.
6. Делахей П., Двойной слой и кинетика электродных процессов ,»Мир», 1967г.
7. Плесков Ю.В. , Фидиновский В.Ю., Вращающийся дисковый электрод, М-Л., «Наука», 1972г.
8. Измайлов Н.А., Электрохимия растворов, М-Л.,»Химия»,1966г.
9. Корыта И., Дворжак И.,Богачкова В., Электрохимия ,»Мир», 1977г.
10. Кравцов В.И. ,Электродные процессы в растворах комплексов металлов , Изд-во ЛГУ; 1969г.
11. Мишенко К.П., Полторацкий Г.М.,Термодинамика и строение водных и неводных растворов элекролитов. М-Л., «Химия» 1976г.,1984г.
12. Латимер В., Окислительные состояния элементов, ИЛ, 1954г.
13. Методы измерения в электрохимии, т.1 и 2, Под ред.Э.Егера и А.Залкинда «Мир», 1974г.
14. Использование наглядных пособий, ТСО, вычислительной техники.
Похожие работы

... Основным критерием, характеризующим состояние поверхности металла, является электродный потенциал. Обычно возможность применения анодной защиты для конкретного металла или сплава определяют методом снятия анодных поляризационных кривых. При этом получают следующие данные: а) потенциал коррозии металла в исследуемом растворе; б) протяженность области устойчивой пассивности; в) плотность тока в ...
... который установил, что лишь два из них: №1 – дицианоаурат калия – KAuCN2 и №2,– дают осадки золота хорошего качества. Вывод Эльснера, по существу, задал направление дальнейших исследований в области электрохимического золочения. Проведенный нами анализ работ де Рюольса показал, что его основная заслуга состоит в том, что он впервые осуществил чисто гальванический процесс. Иными словами, именно ...
... развития гальванотехники в XIX – XX вв. в значительной степени остаётся открытым. Представляется, что его можно решить на основании реконструкции процесса создания гальванического производства; прослеживания, каким областям науки и техники, их конкретным достижениям обязано оно своим становлением; рассмотрения социально-экономических предпосылок возникновения и становления гальванотехники. ...
... Затем детали промывают в горячей проточной воде, производят промывку и активацию. В ванне каскадной промывки происходит противоточная двухступенчатая холодная промывка. Для осаждения цинковых покрытий применяют различные электролиты: кислые, цианистые, аммиакатные, цинкатные и др. В аммиакатном электролите цинк находится в виде комплексных катионов. Аммиакатные соединения цинка получаются при ...
0 комментариев