Навигация
2.3. Методики анализа
2.3.1. Определение содержания Fe(II) при помощи количественного анализа.[81, 82]
К 1г ФМЖ прибавлялся 1мл толуола, 2мл концентрированной HCl и смесь нагревалась на кипящей водяной бане в течение 5 мин, после чего к ней добавлялось 10мл воды, 0,5 мл концентрированной H2SO4 и нагревание продолжалось еще 5 мин. Затем к смеси добавлялись 1мл концентрированной H3PO4, 4мл 5%-ного раствора MnSO4, 1мл толуола и 10мл гексана. Содержимое стакана переносилось в делительную воронку, в которой находилось 20мл воды, и после взбалтывания и отстаивания смеси нижний слой сливался в коническую колбу емкостью 100мл. В воронку повторно добавлялось 10мл воды и после встряхивания и отстаивания нижний водный слой добавлялся к полученному ранее.
Раствор в колбе титровался 0,1н раствором KMnO4 до появления розовой окраски. Параллельно проводился контрольный опыт. 1мл 0,1н раствора KMnO4 соответствует 0,005585г Fe2+.
2.3.2. Определение содержания Fe/III/ при помощи количественного анализа.[81, 82]
К 1г ФМЖ прибавлялся 1мл толуола, 2мл концентрированной HCl и смесь нагревалась на кипящей водяной бане в течение 5 мин, после чего к ней добавлялось 10мл воды, 0,5 мл концентрированной H2SO4 и нагревание продолжалось еще 5 мин. Затем к смеси добавлялись 4мл 5%-ного раствора MnSO4, 1мл толуола и 10мл гексана. Содержимое стакана переносилось в делительную воронку, в которой находилось 20мл воды, и после взбалтывания и отстаивания смеси нижний слой сливался в коническую колбу емкостью 100мл. В воронку повторно добавлялось 10мл воды и после встряхивания и отстаивания нижний водный слой добавлялся к полученному ранее. К пробе перед титрованием добавлялось 2г твердого KJ.
Раствор в колбе титровался 0,1н раствором Na2S2O3, в качестве индикатора использовался крахмал. Параллельно проводился контрольный опыт. 1мл 0,1н раствора Na2S2O3 соответствует 0,005585г Fe3+.
2.3.3. Определение содержания Fe/II/ и Fe/III/ в осадке, образующемся при соосаждении гидроксидов при помощи количественного анализа.[81, 82]
К 1г осадка, просушенного на воздухе при комнатной температуре или отжатого на фильтровальной бумаге, прибавлялось последовательно 2мл концентрированной HCl, 10мл воды, 0,5 мл концентрированной H2SO4, 4мл 5%-ного раствора MnSO4, а для определения Fe/II/ еще и 1мл концентрированной H3PO4. После растворения осадка раствор для определения Fe/II/ титровался 0,1н раствором KMnO4 до появления розовой окраски; к раствору для определения Fe/III/ добавлялось 2г твердого KJ и он оттитровывался 0,1н раствором Na2S2O3 с использованием крахмала в качестве индикатора.
Таким же образом анализировались образцы, стабилизированные минеральными кислотами.
2.3.4. Упрощенный метод определения поверхности по адсорбции воздуха.[84]
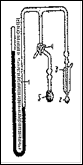
Рис.8. Прибор для определения удельной поверхности.
Прибор для определения удельной поверхности (рис.8) состоит из ртутного манометра 1 со шкалой, длина которого больше 80 см. Одна трубка манометра запаивается под вакуумом, а к другой присоединяется на шлифе ампула 2 с навеской образца. Трубка между шлифом и ампулой заключена в вакуумную рубашку 3, которая позволяет поддерживать постоянным охлаждаемый объем при погружении ампулы в жидкий азот. От трубки, соединяющей ампулу с манометром, сделан отвод с трехходовым краном 4. Вторая трубка от крана сообщается с атмосферой, а к третьей присоединена на шлифе ампула 5, содержащая несколько граммов активного угля.
Для определения удельной поверхности навеска образца помещается в ампулу, которая присоединяется к прибору. Ампула с углем соединяется с манометром и погружается в жидкий азот. После того, как весь воздух из прибора адсорбируется на угле, жидкий азот убирается и начинается десорбция газов в объем. Когда давление в манометре достигает 100—250 мм, поворотом крана ампула с углем отключается от манометра и соединяется с атмосферой. Давление газа в манометре измеряется по шкале с точностью ±0.5 мм. Ампула с образцом погружается в жидкий азот и через несколько минут определяется новое установившееся давление. Поверхность образца определяется по формуле:
![]() , где
, где
S – удельная поверхность,
Δp = p0 - p1
Δp0 = a0 + b0p1 (определяется по коллибровочному графику)
![]()
![]()
p0 и p1 – показания манометра до и после адсорбции на образце.
a, a0, b, b0, S0 – константы прибора.
Измерение адсорбции предлагаемым упрошенным методом связано с некоторыми допущениями, вносящими ошибки. Не учитывается изменение температуры жидкого азота; расчет поверхности проводится в предположении, что адсорбируется чистый азот. Между тем в воздухе неизбежно присутствует кислород. Поверхность измеряется без предварительной откачки образцов. Во многих случаях это не имеет значения, но для некоторых веществ требуется предварительный прогрев образцов для удаления адсорбированной воды.
2.3.5.Электронномикроскопическое исследование.
Препарирование образцов проводилось 2мя способами:
а) нанесения стеклянной палочкой порошка на медные опорные сеточки, расположенные на поверхности стекла и покрытые тонкой коллодиевой пленкой.
б) методом нанесения капли очень разбавленной жидкости на медные опорные сеточки, расположенные на поверхности стекла и покрытые тонкой коллодиевой пленкой.
На поверхность образцов, полученных обоими способами напылялась пленка спектрально чистого углерода толщиной 150-200 Å (вакуумный пост ВУП-4, вакуум 10-4 мм), служащая в качестве подложки при просмотре образца в электронном микроскопе.
Просмотр образцов проводился в электронном микроскопе ЭВМ – 100ЛМ. Количественная обработка результатов выполнялась по полученным ЭМ-снимкам путем определения среднего размера частиц и анализа поверхностных концентраций наблюдаемых частиц. Для количественного анализа подбирались сходные по структуре участки образцов, исследовались не менее трех участков каждого образца.
2.3.6. Рентгенографическое исследование.
Рентгенограммы образцов записывали на рентгеновском дифрактометре HZG-4A (CoKα– излучение). Расшифровка рентгенограмм велась по стандартной методике и идентифицировалась по набору межплоскостных расстояний.
Похожие работы
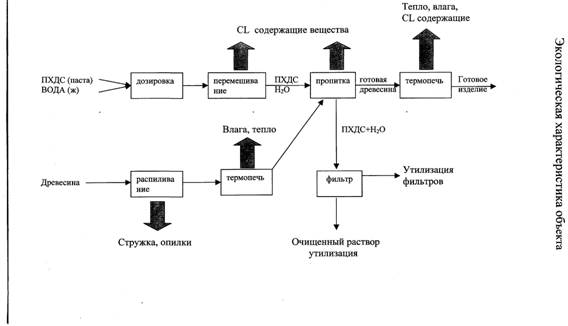
... 4,5. Через краны - бпаста и вода попадают в перемешивающее устройство - 7. По окончании времени перемешивании смесь веществ с помощью крана - 8 попадает в пропиточную ванну - 9, в которую по ленточному конвейеру - 10 поступает древесина из термообрабатывающей печи - 11. После пропитки древесины в течении 30 минут образцы по ленточному конвейеру поступают в печь для последующей сушки. После этого ...
... и, конечно же, за многими другими, которые будут получены, — будущее. В этом направлении и работают многие НИИ и исследователи. Аспекты поиска новых лекарств, изыскание новых лекарственных веществ состоит из трех основных этапов: химический синтез, установление фармакологической активности и безвредности (токсичности). Такая стратегия поиска с большой затратой времени, реактивов, животных, труда ...
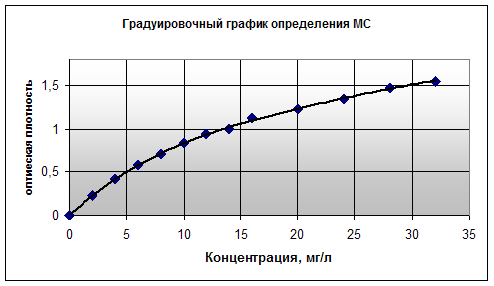

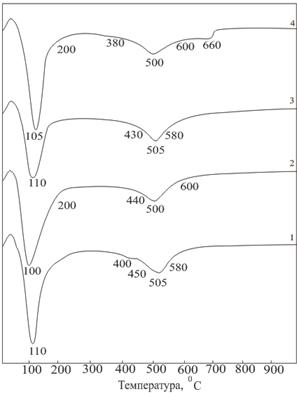

... химическое, макроструктурное модифицирование и одновременное обогащение бентопорошка, позволяют повысить сорбционные свойства и качество готовой продукции. 3.4 Разработка полимерных композиционных материалов на основе органоглин на основе бентонита месторождения «Герпегеж» Объектами исследований в данной части работы являются нанокомпозиты, полученные на основе органомодифицированных ...
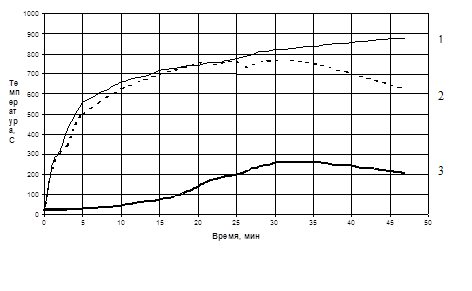

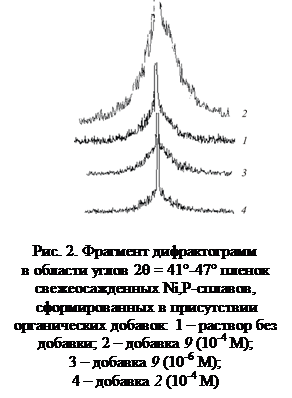
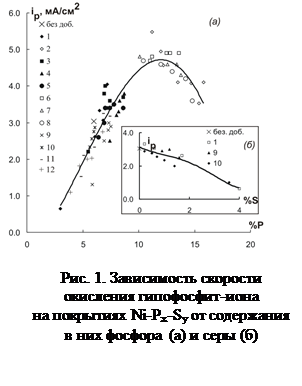
... – x)Na+}xNa+ Таким образом, получены водорастворимые производные фуллерена С60, которые могут быть использованы в химии и химической технологии. УДК 541.138 СТРУКТУРА И СВОЙСТВА НИКЕЛЕВЫХ СПЛАВОВ, МОДИФИЦИРОВАННЫХ ОРГАНИЧЕСКИМИ ДОБАВКАМИ О.В. Долгих, Н.В. Соцкая, Д.В. Крыльский, М.Ю. Хазель Воронежский государственный университет Сплавы никеля уже давно нашли широкое применение в ...
0 комментариев