Навигация
Атомно-адсорбционный спектрохимический анализ тяжелых металлов в почве
43047
знаков
8
таблиц
2
изображения
«АТОМНО-АДСОРБЦИОННЫЙ СПЕКТРОХИМИЧЕСКИЙ АНАЛИЗ ТЯЖЕЛЫХ МЕТАЛЛОВ В ПОЧВЕ»
СОДЕРЖАНИЕ
Введение
1. Атомно-адсорбционная спектрометрия
2. Примеры использования метода в анализе почв
3. Современное оборудование
Литература
ВВЕДЕНИЕ
Экспрессные методы АСА широко применяются в промышленности, сельском хозяйстве, геологии и многих др. областях народного хозяйства и науки. Значительную роль АСА играет в атомной технике, производстве чистых полупроводниковых материалов, сверхпроводников и т. д. Методами АСА выполняется более 3/4 всех анализов в металлургии. С помощью квантометров проводят оперативный (в течение 2—3 мин) контроль в ходе плавки в мартеновском и конвертерном производствах. В геологии и геологической разведке для оценки месторождений производят около 8 млн. анализов в год. АСА применяется для охраны окружающей среды и анализа почв, в криминалистике и медицине, геологии морского дна и исследовании состава верхних слоев атмосферы, при разделении изотопов и определении возраста и состава геологических и археологических объектов и т. д.
1. АТОМНО-АБСОБЦИОННАЯ СПЕКТРОМЕТРИЯ
Метод основан на поглощении ультрафиолетового или видимого излучения атомами газов. Чтобы провести пробу в газообразное атомное состояние, ее впрыскивают в пламя. В качестве источника излучения применяют лампу с полым катодом из определяемого металла. Интервал длин волн спектральной линии, испускаемой источником света, и линии поглощения того же самого элемента в пламени очень узок, поэтому мешающее поглощение других элементов практически не сказывается на результатах анализа.
Существенным отличием атомной абсорбции от пламенно-эмиссионной спектрометрии является то, что в последнем методе измеряется излучение, испускаемое атомами в возбужденном состоянии в пламени, а атомная абсорбция основана на измерении излучения, поглощенного нейтральными, невозбужденными атомами, находящимися в пламени, которых в пламени во много раз больше, чем возбужденных. Этим объясняется высокая чувствительность метода при определении элементов, имеющих высокую энергию возбуждения, т. е. трудно возбуждающихся.
Источником света в ААС служит лампа с полым катодом, испускающая свет, имеющий очень узкий интервал длин волн, порядка 0, 001нм. Линия поглощения определяемого элемента несколько шире испускаемой полосы, что позволяет измерять линию поглощения в ее максимуме. Прибор содержит необходимый набор ламп, каждая лампа предназначается для определения только одного какого-либо элемента.
«Кюветой» в ААС служит само пламя. Поскольку в ААС соблюдается закон Бэра, чувствительность метода зависит от длины поглощающего слоя пламени, которая должна быть постоянной и достаточно большой.
Источники пламени. Применяют пламя, для получения которого в качестве горючего используют ацетилен, пропан или водород, а в качестве окислителя - воздух, кислород или оксид азота (1). Выбранная газовая смесь определяет температуру пламени. Воздушно-ацетиленовое пламя и воздушно-пропановое имеют низкую температуру (2200-2400 ОС). Такое пламя используют для определения элементов, соединения которых легко разлагаются при этих температурах. Воздушно-пропановое пламя используют тогда, когда имеются затруднения в получении ацетилена; такая замена осложняет работу, поскольку в техническом пропане имеются примеси, загрязняющие пламя. При определении элементов, образующих труднодиссоциирующие соединения используют высокотемпературное пламя (3000-3200 О,С) , создаваемое смесью оксид азота (1) - ацетилен. Такое пламя необходимо при определении алюминия, бериллия, кремния, ванадия и молибдена. Для определения мышьяка и селена, превращенных в их гидриды, требуется восстановительное пламя, образующееся сжиганием водорода в аргоно-воздушной смеси. Ртуть определяют (беспламенным методом», поскольку она может существовать в парообразном состоянии и при комнатной температуре.
Мешающие влияния. Мешающих влияний при использовании атомно-абсорбционной спектрометрии немного, проявляются они редко, что и является одним из главных преимуществ этого метода. [1-3]
Преимущества и недостатки методов атомной спектроскопии
Сравнительная оценка возможностей и характеристик различных оптических методов не может носить абсолютного характера в связи с большим разнообразием и спецификой задач анализа. Различными могут быть требования к концентрационному диапазону, точности и нижним границам количественных определений. В зависимости от массы анализируемой пробы существенно различны требования к характеристике пределов обнаружения, достигаемых применяемым методом анализа. Так, располагая большой массой пробы, можно решить задачу определения микропримесей с помощью методов анализа, характеризуемых низкими относительными пределами обнаружения. Если же в распоряжении аналитика имеется лишь малая масса пробы, метод анализа должен характеризоваться низкими абсолютными пределами обнаружения интересующих элементов-примесей. Не последнюю роль в оценке недостатков и достоинств различных методов играет экономичность этих методов: стоимость аппаратуры, расход энергии, трудовые затраты, продолжительность анализа.
Атомно-эмиссионный спектральный анализ - практически самый распространенный экспрессный высокочувствительный метод идентификации и количественного определения малых содержаний элементов. Важным достоинством метода по сравнению с другими оптическими спектральными, а также многими химическими и физико-химическими методами анализа является возможность одновременного количественного определения большого числа элементов в широком интервале концентраций с приемлемой точностью при использовании малой массы пробы.
Достоинствами метода атомно-флуоресцентного анализа являются сравнительно низкий уровень фона, высокая селективность измерений, малые спектральные помехи, что позволяет детектировать слабые аналитические сигналы и соответственно очень малые абсолютные количества элементов. К недостаткам метода атомно-абсорбционной и в определенной мере атомно-флуоресцентной спектрометрии следует отнести затруднительность одновременного определения нескольких элементов.
С точки зрения возможности определения ультрамалых абсолютных содержаний элементов-примесей (#10-11-10-12 г) из оптических атомно-спектральных методов заслуживают особого внимания новые атомно-флуоресцентные и атомно-ионизационные методы с возбуждением и ионизацией атомов с помощью перестраиваемых лазеров на красителях, а также некоторые современные варианты оптических атомно-эмиссионного и атомно-абсорбционного методов анализа. В последнее время широкое распространение получил атомно-эмиссионный анализ с возбуждением спектров в высокостабильной индуктивно-связанной плазме (ИСП-АЭС). Современные анализаторы на основе этого метода обычно включают полихроматор с решеткой и приемники с зарядовой связью. Такая оптическая схема позволяет одновременно регистрировать все спектральные линии в ультрафиолетовом и видимом диапазонах. Программное обеспечение современных ИСП-АЭС-анализаторов способно автоматически рассчитывать концентрацию определяемых элементов по интенсивности их спектральных линий с коррекцией фона и возможных спектральных наложений. Соответственно такие анализаторы отличаются высокой точностью и продуктивностью.[3-7]
2. ПРИМЕРЫ ИСПОЛЬЗОВАНИЯ МЕТОДА В АНАЛИЗЕ ПОЧВ Определение тяжелых металлов атомно-абсорбционным методомМетодика предназначена для выполнения измерений массовой концентрации металлов (марганца, меди, железа, цинка, молибдена) в пробах природных и сточных вод атомно-абсорбционным методом с электротермической атомизацией с использованием атомно-абсорбционного спектрометра "МГА-915".
Таблица 1
Диапазон измеряемых концентраций в пробах воды без разбавления:
| Элемент | Концентрация |
| Цинк | 0,1 - 8,0 мг/дм3 |
| Хром | 0,001 - 0,1 мг/дм3 |
| Марганец | 0,0003 - 0,050 мг/дм3 |
| Медь | 0,0005 - 0,070 мг/дм3 |
| Железо | 0,005 - 0,060 мг/дм3 |
Объем дозируемой в атомизатор пробы - от 5 до 40 мм3.
При анализе проб воды с массовой концентрацией металлов, превышающей верхнюю границу диапазона, допускается разбавление пробы бидистиллированной (деионизованной) водой, но не более чем в 100 раз. (Zn, Cr), не более чем в 1000 раз (Mn, Cu, Fe)
Метод измеренияМетод измерения основан на резонансном поглощении света свободными атомами металлов, возникающем при пропускании света через слой атомного пара в графитовой печи атомно-абсорбционного спектрометра "МГА-915". Содержание металлов определяется величиной интегрального аналитического сигнала и рассчитывается по предварительно установленной градуировочной зависимости.
Характеристика погрешности измеренияМетодика обеспечивает выполнение измерений с погрешностью, не превышающей величин, указанных в табл.2.
Таблица 2Значения характеристики погрешности измерений для доверительной вероятности Р=0,95
| Определяемый элемент | Воды природные | |
| Диапазон измеряемых массовых концентраций, мг/дм3 | Характеристика погрешности измерений, ±δ, % | |
| Марганец | 0,0003-0,005 | 35 |
| Медь | 0,0005-0,010 | 50 |
| Цинк | 0,1-0,4 | 35 |
| Железо | 0,005-0,010 | 35 |
| Хром | 0,001-0,005 | 40 |
| Спектрометр атомно-абсорбционный "МГА-915" | ТУ 4434-915-205016233-98 |
| Весы лабораторные общего назначения 2-го класса точности, например ВЛР-200 | ГОСТ 24104 – 88 |
| Меры массы | ГОСТ 7328 – 82 |
| Мерные колбы 2-1000-2, 2-100-2, | ГОСТ 1770 – 74 |
| Пипетки градуированные 2-го класса точности вместимостью 1, 2 , 5 и 10 см3 | ГОСТ 29227 - 91 |
| Дозатор пипеточный одноканальный переменного объема 5-50 мм3. Погрешность измерения – не более ±5 % | ТУ 9452-001-33189998-95 |
| Государственные стандартные образцы состава раствора определяемых ионов (1 мг/см3, погрешность аттестованного значения ±1 %): | ГСО 7266 - 96 |
| • марганца | ГСО 7266 - 96 |
| • меди | ГСО 7255 - 96 |
| • железа | ГСО 7254 - 96 |
| • цинка | ГСО 7256 - 96 |
| • хром | ГСО 7768 - 2000 |
Допускается использование средств измерений и стандартных образцов с аналогичными или лучшими метрологическими характеристиками. Средства измерений должны быть поверены в установленные сроки.
Отбор и хранение пробОбщие требования к отбору проб по ГОСТ Р 51592-2000. Отбор проб природной воды производится по ГОСТ 17.1.5.05-85, сточной воды по НВН 33.5.3.01-85. Объем отбираемой пробы составляет не менее 50 см3.К пробе добавляют 3 см3 концентрированной азотной кислоты на 1 дм3 пробы и хранят в посуде из полиэтилена, полипропилена или фторопласта. Пробы перед анализом фильтруют через бумажный фильтр "белая лента" или пористый фильтр с диаметром пор 0,45 мкм. При фильтровании первые порции фильтрата (не менее 5 см3) следует отбросить. Посуду, предназначенную для отбора проб и хранения проб, промывают раствором азотной кислоты, а затем дистиллированной и бидистиллированной (ионизированной) водой. Срок хранения законсервированной пробы - 3 дня.
Подготовка к выполнению измеренийДля каждого раствора необходимо использовать свою пипетку. Раствор из колбы наливают в стаканчик и из него набирают пипетку. Запрещается погружать пипетку во весь объем раствора во избежание загрязнения.
Рекомендуется иметь отдельный набор посуды для приготовления растворов каждого элемента.
Приготовление растворов
Все растворы готовят на бидистиллированной (деионизованной) воде.
Раствор азотной кислоты, объемная доля 2%
В коническую колбу помещают 300-400 см3 бидистиллированной деионизованной воды, осторожно, при перемешивании вливают 10 см3 концентрированной азотной кислоты, доводят до 500 см3 бидистиллированной водой и перемешивают. Раствор устойчив при хранении в закрытом сосуде из полиэтилена, полипропилена или фторопласта длительное время. Контроль качества раствора азотной кислоты проводят ежедневно перед началом работы. Процедура контроля состоит в измерении массовой концентрации элементов в соответствии с п.7 методики. Если измеренное значение превышает 30% нижней границы диапазона измерения элемента (п.1),то раствор необходимо приготовить заново.
Приготовление градуировочных растворов
Приготовление рабочих растворов массовой концентрации 100мг/дм3
В мерную колбу вместимостью 50 см3 помещают при помощи пипетки 5 см3 государственного стандартного образца состава раствора соответствующего иона, доводят до метки 2 % -ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течение 1 месяца.
Приготовление рабочих растворов массовой концентрации 2мг/дм3
В мерную колбу вместимостью 100 см3 помещают при помощи пипетки 2 см3 рабочего раствора массовой концентрации 100 мг/дм3, приготовленного по п. 6.2.2.1, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течении 2 недель.
Приготовление рабочих растворов массовой концентрации 1мг/дм3
В мерную колбу вместимостью 100 см3 помещают при помощи пипетки 1 см3 рабочего раствора массовой концентрации 100 мг/дм3, приготовленного по п. 6.2.2.1, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор устойчив при хранении в полиэтиленовой посуде в течении 2 недель.
Приготовление рабочих растворов массовой концентрации 20мкг/дм3
В мерную колбу вместимостью 100 см3 помещают при помощи пипетки 2 см3 рабочего раствора массовой концентрации 1 мг/дм3, приготовленного по п. 6.2.2.3, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Приготовление рабочих растворов массовой концентрации 10мкг/дм3
В мерную колбу вместимостью 100 см3 помещают при помощи пипетки 1 см3 рабочего раствора массовой концентрации 1 мг/дм3, приготовленного по п. 6.2.2.3, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Приготовление рабочих растворов массовой концентрации 1мкг/дм3
В мерную колбу вместимостью 50 см3 помещают при помощи пипетки 5 см3 рабочего раствора массовой концентрации 10 мкг/дм3, приготовленного по п. 6.2.2.5, доводят до метки 2%-ным раствором азотной кислоты и перемешивают. Раствор используют свежеприготовленным.
Градуировка спектрометра
Для построения градуировочной зависимости аналитического сигнала от массы элемента в графитовую печь атомизатора вводят дозатором необходимый объем (от 5 до 40 мм3) градуировочных растворов соответствующего элемента. Диапазоны построения градуировочной зависимости приведены в таблице 2. Необходимо использовать не менее 5 точек в указанном в таблице 2 диапазоне массы. При построении градуировочной зависимости следует начинать с меньших значений массы элемента и от них переходить к более высоким. В таблице 3 приведены рекомендуемые для внесения объемы градуировочных растворов.
Рекомендуемые режимы обработки градуировочных растворов и проб приведены в таблице 4.
Измерение с каждой массой элемента проводят 5 раз в соответствии с Руководством по эксплуатации спектрометра (далее РЭ) и рассчитывают среднее арифметическое значение полученных значений. Затем запускают процедуру «Ручная градуировка» и вводят с клавиатуры компьютера массу элемента (в пиктограммах) и соответствующие им величины средних значений аналитического сигнала. Полученную градуировочную зависимость можно просмотреть в режиме «Градуировка»/«Просмотр».
Таблица 3
Диапазоны построения градуировочных зависимостей.
| Элемент | Диапазон измерения, мг/дм3 | Диапазон масс, пг |
| Марганец | От 0.0003 до 0.050 включительно | 10 - 400 |
| Медь | От 0.0005 до 0.070 включительно | 20 - 600 |
| Железо | От 0.005 до 0.060 включительно | 40 - 600 |
| Цинк | От 0.1 до 8.0 включительно | 5000 - 80000 |
| Хром | От 0.001 до 0.1 включительно | 40 - 600 |
Таблица 4
Рекомендуемые способы внесения элемента в атомизатор
| Масса, пг | Концентрация градуировочного раствора, мкг/дм3 | Объем градуировочного раствора, мм3 |
| 10 | 1.0 | 10 |
| 20 | 1.0 | 20 |
| 40 | 1.0 | 40 |
| 100 | 10.0 | 10 |
| 200 | 10.0 | 20 |
| 400 | 10.0 | 40 |
| 600 | 20.0 | 30 |
| 800 | 20.0 | 40 |
При высоких значениях массы элемента может наблюдаться отклонение градуировочной зависимости от линейной. В этом случае рекомендуется ограничиться более узким, чем в указано в табл. 2 интервалом массы определяемого элемента для построения градуировочной зависимости.
Контроль стабильности градуировочной зависимости
Контроль стабильности градуировочной зависимости состоит в проведении не менее двух параллельных измерений массовой концентрации растворов, заново приготовленных по п. 6.2.2, перед началом работы, и после анализа 15-20 проб.
Градуировка признается стабильной, если расхождение между заданным и измеренным значением концентраций не превышает 15% от заданного значения. В этом случае процесс измерений признается подконтрольным, и результаты измерений массовой концентрации элемента в пробах за период между двумя последовательными процедурами контроля стабильности градуировочной характеристики принимаются в качестве окончательных результатов.
При несоответствии полученных результатов указанному нормативу процесс градуировки необходимо повторить.
Выполнение измеренийВводят дозатором в графитовую печь атомизатора от 5 до 40 мм3 анализируемой пробы (в зависимости от ожидаемого содержания) и производят измерение в соответствии с выбранным режимом работы (таб. 4). Режимы при измерении градуировочных растворов и проб (за исключением стадии пиролиза) должны совпадать. Температура и продолжительность пиролиза зависят в первую очередь от матричного состава пробы. При анализе сравнительно чистых или разбавленных сточных вод режим пиролиза можно не использовать.
Порядок проведения измерений осуществляется в соответствии с Руководством по эксплуатации спектрометра. Объем дозированной пробы вводится с клавиатуры компьютера по запросу программы. После завершения измерения на дисплей компьютера выводится величина интегрального аналитического сигнала, масса и концентрация определяемого компонента. Полученные данные автоматически протоколируются. Анализ пробы осуществляется минимум 2 раза.
Если измеренное значение массы элемента в пробе выходит за область линейности градуировочной характеристики, то пробу необходимо разбавить бидистиллированной (деионизованной) водой, предварительно проверенной на наличие примеси определяемого элемента. Затем разбавленную пробу анализируют как описано выше. Коэффициент разбавления пробы Q вычисляют по формуле[8-16]
|
| (1) | |
| где Vк | объем разбавленной пробы, см3; | |
| Vа | аликвотная порция исходной пробы, взятая для разбавления, см3. | |
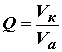
Разработка методического обеспечения атомного оптического спектрального анализа почв и биологических материалов для экологической экспертизы на токсичные металлы[23]
Разработан простой и экспрессный способ кислотного разложения почв и биологических объектов при воздействии ультразвуком для определения ртути, свинца и других тяжелых металлов из одного раствора. На этой основе предложены новые экспрессные методики последовательного атомно-абсорбционного определения ртути на ртутном анализаторе “Юлия-2” и свинца, цинка, меди – на пламенном атомно-абсорбционном спектрометре “AAS-1“ в пламени пропан-воздух.
Проблему повышения чувствительности определения свинца и многоэлементного анализа решали спектрографическим методом на дифракционном спектрографе ДФС 8–1 после упаривания раствора пробы с углеродным коллектором и введением сухого концентрата в дуговой разряд воздушной струей на спектральной установке “Полюс –2”.
Методики были применены к анализу почв, донных отложений и волос человека.
Было изучено распределение Hg , Pb, Zn , Cu в почвах и в выделенных из них гуминовых кислотах по разрезу учебного полигона ИГУ, разрабатываемого совместными усилиями преподавателей и студентов биолого-почвенного, географического и химического факультетов.
Проведена экспертиза почв Усольского района, находящихся под несанкционированной свалкой УПО “Химпром”, и было выявлено загрязнение по натрию, бору и хрому.
Представлены содержания Hg , Pb и Zn в волосах детей 6 и 12 лет Академгородка г.Иркутска, которые были исследованы с целью медико-экологического мониторинга в рамках довузовской программы со школьниками “Экология и здоровье”.
Выполнен аттестационный анализ разработанных в Институте геохимии СО РАН стандартных образцов состава донных отложений оз. Байкал БИЛ–1 и БИЛ–2 на ртуть. Определены содержания исследуемых металлов в донных отложениях Братского водохранилища. Подтверждены данные о повышении содержания ртути (ртутная проблема водохранилищ).
Квантовохимическое моделирование и исследование пиролиза серусодержащих хелатов меди, кадмия свинца атомно-абсорбционным методом[22]
Серусодержащие комплексообразующие модификаторы матрицы позволяют устранить влияние основы, улучшить воспроизводимость и снизить предел обнаружения при атомно-абсорбционном (АА) определении меди, кадмия, свинца и др. элементов в сложных объектах. Эффективность добавки определяется высокотемпературным процессом пиролиза комплексов в графитовых печах на доатомизационных станциях. АА методом установлено влияние на аналитический сигнал свинца (II), меди (II) и кадмия (II) добавок диэтилдитиокарбамата натрия и бензтиазолилмеркаптометилсульфида. Вышеуказанные хелаты были выделены в твердом виде, подтвержден их состав и дериватографическим методом установлено, что их разложение носит ступенчатый характер и завершается к 500 С. Квантовохимическое моделирование позволило оценить вероятный путь пиролиза хелатов. Поиск оптимальной конфигурации ядерного остова выполнялся в приближении Девидона-Флетчера. расчет электронной структуры выполнен в полуэмпирическом приближении МО ЛКАО. Использован комплекс квантовохимических программ МОРАС 6. Для диэтилдитиокарбамата свинца с точки зрения величины теплоты возможного процесса наиболее вероятным представляется радикальный пиролиз с разрывом связи Pb–S и образованием связанного с сухим углеродным остатком свободного радикала, стабилизированного системами конденсированных колец образующихся при карбонизации углеродсодержащих соединений. Другими продуктами пиролиза являются промежуточный хелат с меньшим, по сравнению с исходным, содержанием диэтилдитиокарбамата и, вероятно, сероводород. Аналогично протекает пиролиз и бензтиазолилмеркаптометилсульфида меди. Диэтилдитиокарбамат кадмия при разложении также образует свободный радикал, сульфид кадмия и газообразные продукты, состав которых существенно изменяется в зависимости от условий нагрева атомизатора, содержания кислорода в инертном газе, от состояния поверхности печи и т.д.
Предложенные модели пиролиза экспериментально подтверждены дериватографическим, рентгенофазовым методами, ЭПР-спектроскопией, исследованием молекулярных спектров АА методом. Обнаруженная общая стадия высокотемпературного разложения серусодержащих комплексов с образованием свободных радикалов изменяет функцию испарения и переноса металлов в графитовой печи, что способствует термостабилизации и объясняет изменение метрологических характеристик АА методик в присутствии добавки.
Изучение поведения токсичных элементов в природных средах методом атомной абсорбции.[21]
Проведено комплексное исследование загрязнения воздушного пространства, снега, почв, водных источников на основе атомно-абсорбционного определения токсичных элементов в каменных углях, золе уноса Беловского района Кемеровской области.
В качестве базовой информации были использованы результаты аналитических исследований 100 образцов твёрдых веществ в снежном покрове в период максимального влагозапаса, твёрдых веществ, содержащихся в приземном воздухе, 48 образцов воды из питьевых источников, 130 образцов почв, 30 образцов листьев древесных растений, 70 образцов растительных кормов сельскохозяйственных животных, 120 образцов пищевых продуктов человека. Всего было выполнено более 9000 элементо-определений с целью обнаружения в них Cu, Be, Hg, Zn, Cd, Pb, As, Cr, Mn, Fe, Co, Ni.
Определение химических элементов проводили на спектрофотометрах Сатурн-2М и ААS-1N, с пределами обнаружения в мкг/мл: 0,002 для Ве и Аs; 005 для Mn и Cu; 0,01 – Zn, Cd ,Cr; 0,02 – Ni; 0,05 – Cr; 0,1 для Hg.
Осуществлён анализ проблемы загрязнений воздушного бассейна теплоэнергетическими объектами. Обосновано приоритетное значение твёрдых атмосферных выбросов теплоэнергетических объектов для местных экосистем и популяций человека. Изучен состав токсичных химических веществ в кузбасских каменных углях и рассчитан их баланс в процессе сжигания твёрдого топлива на Беловской ГРЭС. Установлено, что на ГРЭС с золой уноса в воздушный бассейн поступает 3-39% массы токсичных веществ, а 61-97 % этих веществ ассоциируется с золошлаковой смесью и сбрасывается в золоотвал. Техногенная химическая нагрузка на изучаемую территорию определена по содержанию вредных элементов в снежном покрове в период максимального влагозапаса. Эти данные использованы для расчёта доз химических токсикантов, поступающих в организм местных жителей.
Определение форм тяжелых металлов в снежном покрове после экстракции тиопирином[20]
В работе представлены экспериментальные результаты снегосъемки 2002-2003 гг. на опорных площадках промышленного центра и в области его влияния. Методом атомно-абсорбционного анализа водной и кислотной вытяжек из почвогрунтов, а также экстрактов из твердой компоненты снега и образцов после их мокрого озоления количественно определены различные формы свинца. В работе было показано, что оптимальное количество тиопирина для извлечения катионов металлов составляло 4*10-3 моль при фиксированном количестве трихлоруксусной кислоты 2*10-2 моль.Тиопирин хорошо растворим в воде при нагревании. Данный реагент склонен к протонизации с образованием катионной формы: Тиопирин и его аналоги обладают сочетанием ярко выраженных свойств ароматических соединений и алифатических аминов. Это объясняется наличием в гетероцикле секстета p - электронов, т.к. в молекуле тиопирина неподеленные p - электроны атомов азота включаются в общую p - электронную систему тиопиринового цикла. Вследствие этого атомы азота теряют свои электрондонорные свойства, а атом серы получает значительный отрицательный заряд. Таким образом, донорным атомом служит атом серы. Тиопирин является монодентным лигандом, взаимодействующим с неорганическими катионами Pb(II), хемосорбированными на частицах твердого аэрозоля и пылевых выпадений в виде неорганических солей с анионом X 2 - ( сульфид, сульфат, карбонат). При этом образование комплекса со свинцом зависит от pH
C11H12N2S + PbX +2H+ + 2CCl3COO - « [C11H12N2SPb] (CCl3COO)2 + HX- + H+
При этом протоны водорода могут встраиваться во внутреннюю сферу комплекса.
Таким образом установлено, что на поверхности твердых частиц снежной массы преобладает на 80 - 90% неорганический свинец, на органические формы свинца приходится 3 - 9 %, в том числе 2 - 5 % антропогенного происхождения. Метод определения соотношения форм предлагается использовать для экологического контроля за состоянием атмосферы и снежного покрова в зимний лей.
Спектрометрические характеристики растворов гумуса, исследование их комплексообразования с металлами[19]
Исследование комплексообразующих (КО) свойств гуминовых веществ (ГВ) проводят с целью их использования для уменьшения токсичности тяжёлых металлов в природных объектах, что представляет интерес для агрохимии, почвоведения и курортологии. Сложный состав ГВ, большая зависимость их строения от географического положения объекта, из которого они выделены (почвы, уголь, торф, сапропель, вода), не позволяют распространять результаты исследований и методологические подходы ко всем ГВ.
Методом ААС установлено, что в образцах ГВ содержатся: К, Са, Mg от 1 до 10%; Fe, Zn, Na от 10-2 до 10-1%; остальные металлы от 10-4 до 10-3%. Определяющее влияние на процесс КО ГВ с Cu2+ и Pb2+ оказывает присутствие металлов первой группы. Обработка ГВ 0.1н HCl в течение 12 часов ведет к потере К и Са. Использование Н-формы ГВ более перспективно для КО с металлами.
Электронные спектры водных растворов ГВ и их растворов с Cu2+, Pb2+ в ацетатно-аммиачных буферах (при рН 5, 6, 7, 8) показали, что растворы ГВ в воде имеют интенсивные полосы поглощения, например λ1max=207-208 нм, которые в буферных растворах смещаются в длинноволновую область до λ2max=235 нм. КО ГВ с ионами металлов имеет сложный характер. Поэтому для изучения КО были также использованы ИК-спектры. Образование связи карбоксилат-ион-металл устанавливали по исчезновению полосы поглощения 1720 см-1 и появлению полосы 1580 см-1.
На основе полученных данных можно прогнозировать поведение ионов металлов в природных объектах, в том числе при содержаниях на уровне ПДК.
Термическая атомно-абсорбционная спектроскопия как метод диагностики форм нахождения тяжелых металлов в объектах окружающей среды и минералах[18]
Термический атомно-абсорбционный анализ (ТАА) является совмещением термического и атомно-абсорбционного анализа. Метод основывается на одновременной регистрации сигнала абсорбционности от спектрометра и температуры пробы. При этом диагностическими характеристиками являются температурные параметры выхода элементов, зависящие от форм присутствия данных элементов в минеральном веществе. Предварительно производится калибровка этих параметров по синтетическим минералам с заданными формами нахождения элемента. В настоящее время метод разработан для анализа форм ртути, кадмия, свинца, цезия. Данный метод позволяет решать широкий спектр задач геохимии, неорганической химии и охраны окружающей среды.
Проведены системные исследования форм нахождения тяжелых металлов (Hg и Cd) в осадках водохранилищ Ангарского каскада, донных отложений озера Байкал и акватории Охотского, Берингова и Японского морей. Установлено, что основной поток ртути в озеро Байкал и его донные отложения направлен из атмосферы. В донных отложениях Братского водохранилища обнаружено широкое развитие сорбционных форм кадмия, что необходимо учитывать при мониторинге кадмиевого загрязнения на территории Иркутской области, так как он может быть токсичным для органических объектов водохранилищ.
ТАА позволил показать различие в механизмах поглощения микроэлементов при исследовании образования устойчивых форм Cd и Hg в пирротине и галените (в условиях гидротермального синтеза при повышенных температурах и давлениях). Это стало возможным благодаря сопряжению методов ТАА и спектроскопии поверхности. Большое влияние на механизм захвата оказывают дефекты структуры исходных минералов. Особое значение имеет механизм с участием так называемых "неавтономных фаз", так как они более устойчивые по сравнению с поверхностными комплексами.
Похожие работы
... и кондуктометрия. Наиболее эффективными вольтамперометрическими методами являются дифференциальная импульсная полярография (ДИП) и инверсионный электрохимический анализ (ИЭА). Сочетание этих двух методов позволяет проводить определение с очень высокой чувствительностью - приблизительно 10-9 моль/л, аппаратурное оформление при этом несложно, что дает возможность делать анализы в полевых условиях. ...
0 комментариев