Вискозиметрия и кинетика начальных стадий отверждения полиуретанов
Химизм реакций образования полиуретанов исследовали во многих работах, что позволило составить общее представление об этом процессе [1]. Значительно меньше изучено изменение реологических свойств в процессе образования полиуретанов [2, 3], хотя этот вопрос представляет как общетеоретический, так и технологический интерес.
Процесс формирования трехмерных сетчатых структур при образовании полиуретанов (ПУ) из полифункциональных олигомеров можно рассматривать как происходящий в две стадии [2—4]. Вначале интенсивно увеличивается вязкость вследствие удлинения цепей и образования ветвлений при сохранении текучести реакционной массы, затем в критической точке («гель-точке»), когда образуется сплошная трехмерная сетка химических связей, текучесть утрачивается. С точки зрения оценки технологических свойств («перерабатываемое»), наибольший интерес представляет первая стадия, когда отверждаемый ПУ способен формоваться в изделия. В литературе описаны многие результаты изучения кинетики процесса образования ПУ, выполненные различными методами (ИК-спектроскопия, дифференциально-сканирующая калориметрия, торсионный анализ и т. д.) [1]. Однако наиболее прямую информацию относительно влияния протекания химической реакции на свойства образующегося ПУ дает вискозиметрический метод, который позволяет также высказать определенные суждения о макрокинетике процессов образования полимеров, поскольку может быть установлена связь между нею и реокинетикой полимеризации или поликонденсации [5].
В этой связи задача настоящей работы - изучение закономерностей роста вязкости в процессе отверждения ПУ в связи с исследованием кинетики начальной стадии этого процесса.
Исследовали макродиизоционат, синтезированный на основе политетраметилен-гликоля и 2,4-толуилендиизоционата в мольном соотношении 1: 2.Исходный политетраметиленгликоль содержал 3,5% гидроксильных групп. Mw/Mn, определенное гель-хроматографически, равно 1,7; Af„=1020 (эбуллиоскопия). Перед синтезом 2,4-то-луилендиизоционат перегоняли в вакууме (1,33 кПа) при 120°. Синтезированный макродиизоционат анализировали на содержание NCO-групп по известной методике. Отвердителем служил 3,3'-дихлор-4,4'-диаминодифенилметан, двукратно перекристаллизованный из гептана.
Реокинетические исследования проводили на ротационном вискозиметре «Рео-тест-2» с рабочим узлом конус-плоскость при низких скоростях сдвига. Для получения реакционной смеси макродиизоционат смешивали с необходимым количеством предварительно расплавленного 3,3'-дихлор-4,4'-диаминодифенилметана (т. пл. 103,2°) в быстродействующем смесительном устройстве при комнатной температуре. Затем реакционную смесь (~0,1 мл) помещали в рабочий узел вискозиметра, нагретый до температуры опыта. Специально проведенные эксперименты показали, что вследствие небольшого объема навески и интенсивного теплоотвода режим отверждения близок к изотермическому. Отклонения температуры от заданной не превышали 1°. Параллельно при условиях, которые стремились сделать максимально адекватными используемым в реокинетическом эксперименте, проводили отверждение реакционной системы вне вискозиметра с тем, чтобы оценить кинетику процесса: для этого через определенные промежутки времени определяли содержание NCO-групп. Погрешность при определении вязкости не превышала 6%, концентрации NCO-групп 5%. Третьей (кроме вискозиметрической и химической) независимой методикой изучения кинетики отверждения была калориметрия, с помощью которой оценивали скорость тепловыделения в процессе отверждения.
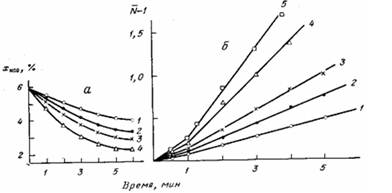
Рис. 1. Изменение концентрации концевых NCO-групп (а) и среднечис-ленной степени поликонденсации (б) в процессе реакции макродиизо-пианата с 3,3'-дихлор-4,4'-диаминодифенилметаном при 60 (1), 70 (2), 80 (3), 90 (4), 100° (5)
Увеличение вязкости на начальных стадиях отверждения ПУ может быть обусловлено удлинением макромолекулярных цепей вследствие протекания процесса линейной конденсации, поскольку используемые олиго-меры представляют собой бифункциональные соединения. В этом случае скорость роста макромолекулярных цепей отражается кинетикой уменьшения концентрации концевых функциональных групп. Изменение концентрации концевых NCO-групп до образования визуально наблюдаемой в растворе нерастворимой фракции показано на рис. 1, а. Кинетика процесса поликонденсации двух бифункциональных мономеров при их экви-мольном соотношении обычно описывается уравнением второго порядка по концентрации функциональных групп, интегрирование которого дает следующее известное выражение для среднечисленной степени поликонденсации образующего полимера: N—l=Xokt, где N — степень поликонденсации; Хо — начальная концентрация функциональных групп; к — константа скорости реакции; t — время.
Результаты обработки экспериментальных данных по этой формуле приведены на рис. 1, б. Однако экспериментальные данные на рис. 1, б аппроксимируются не одной прямой, а двумя линейными участками с различными угловыми коэффициентами. Изменение угла наклона прямой на рис. 1, б может свидетельствовать либо об изменении константы скорости (т. е. в данном случае об ускорении реакции на втором этапе), либо об изменении механизма реакции после нескольких начальных актов конденсации. К аналогичным выводам, свидетельствующим о невозможности описания даже начальных стадий отверждения одним значением константы скорости, приводит также обработка данных изотермической калориметрии.
В предположении второго порядка реакции и пропорциональности скорости превращения £1 интенсивности тепловыделения q зависимость q($) должна иметь вид
![]()
где с — нормирующая приборная константа, связывающая J3 и q и пропорциональная суммарному тепловому эффекту реакции. Константу с следует подбирать так, чтобы наилучшим образом аппроксимировать экспериментальную зависимость q(t) с помощью функции (1). Это проделано на рис. 2, из которого видно, что если такая аппроксимация вполне удовлетворительна для большей части процесса на его второй (заключительной) стадии, то она не годится для первой стадии. Справедливо и обратное. Это означает, что константа к не может быть постоянной для всего процесса.
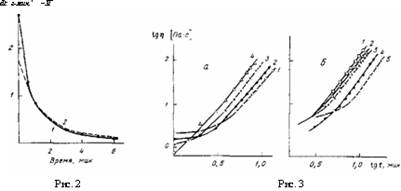
Рис. 2. Изменение скорости тепловыделения д (1) и степени превращения $ (2) в процессе реакции
Рис. 3. Изменение вязкости реакционной массы во времени, а: 60 (1), 70 (2), 80 (3) и 90° (4); б: количества 3,3'-дихлор-4,4'-диаминодифенилметана 1,1 {Т); 1,0 (2); 0,9 (3); 0,8 (4); 0,7 мол.% (5); 80°
Характерно, что точка расхождения экспериментальной и расчетной кривых на рис. 2 совпадает с точкой перелома на рис. 1, б. Полученные результаты, т. е. изменение наклона прямой на рис. 1, б и расхождение кривых на рис. 2, свидетельствуют о том, что в рассматриваемом случае изменяется характер процесса, отражающийся на его кинетике. Попытаемся понять причину этого явления с помощью реокинетического анализа.
Если образование полимера происходит по механизму линейной поликонденсации, то следует ожидать, что вязкость реакционной системы должна изменяться следующим образом [4, 6]: r=K(x0kt)a, где К—константа, зависящая от температуры и природы полимера, т. е. зависимость lg т] от lg t должна быть линейной с угловым коэффициентом, равным 3,4. Соответствующие данные приведены на рис. 3, а. Сопоставление результатов, приведенных на рис. 1 и 3, позволяет построить зависимость вязкости образующегося ПУ от молекулярной массы, которая приведена на рис._4. Полученная зависимость описывается степенной функцией вида т)—Na, где показатель степени а изменяется от 1,0 до 4,6. Если величина а=1,0 является характерной для полимера с небольшой ММ, то значение 4,6 превышает «универсальное» значение этого показателя, равное 3,4. Такое же завышенное значение этого показателя следует и из рис. 3.
Рассмотрим также температурную зависимость вязкости на различных стадиях процесса поликонденсации, что важно, поскольку эта зависимость включает формальные реологические константы материала.
Температурная зависимость вязкости реакционной системы определяется энергией активации как собственно процесса химической реакции U, так и энергией активации вязкого течения Е, и для поликонденсации «эффективные» значения энергии активации выражаются следующим образом [6]: Et=E—all; E=U—(Е/а), где Et —«эффективные» энергии активации, отражающие температурные зависимости вязкости при постоянной продолжительности реакции Et, или времени, необходимого для достижения определенного уровня вязкости £"„.
Результаты обработки экспериментальных данных согласно этим формулам приведены на рис. 5. Сравнение величин энергии активации процесса конденсации U, определенных из зависимости к=к0 ехр (— U/RT) и но рис. 5, при значении а=4,6 и независимо измеренном значении Е= =41,6 кДж/моль дает практически одну и ту же величину, равную 31,5 кДж/моль.

Рис. 4. Зависимость вязкости образующегося ПУ от степени поликопденсации в процессе реакции макродиизоцианата с 3,3'-дихлор-4,4'-диаминодифенилметаном при 60 (1), 70 (2), 80 (3) и 90° (4) в линейных (я) и в двойных логарифмических координатах (б)
Таким образом, полученные результаты (подчинение макрокинетики реакции уравнению второго порядка и характерным для конденсации рео-кинетическим соотношениям) свидетельствуют о том, что в рассматриваемом случае, т. е. при взаимодействии бифункционального макродиизоцио-ната с диамином, действительно протекает процесс поликонденсации до образования визуально наблюдаемой нерастворимой фракции. Однако, как уже отмечено, наблюдали две особенности: во-первых, изменение угла наклона графиков на рис. 1, б, и, во-вторых, аномально высокое значение показателя а в зависимости т]—Na, равное 4,6. По-видимому, эти особенности можно объяснить тем, что после первых актов реакции в растущих цепях появляются разветвления, приводящие к образованию зародышей микрогелей вследствие появления нерастворимости фракции.
Это предположение подтверждается данными, приведенными на рис. 6, где сопоставлены кривые нарастания вязкости, полученные в настоящей работе и в работе [7]. В последнем случае для отверждения ПУ использовали трехфункциональный отвердитель, что заранее предполагает образование разветвленных цепей начиная с первых актов реакции. Аналогия характера зависимостей r(t) и равенство показателей а=4,6 в обоих случаях, свидетельствует об эквивалентности физической картины сопоставляемых процессов, т. е. свидетельствует о том, что и в нашем случае образуются разветвленные продукты.
Существование химических связей, образующих разветвленные макромолекулы при взаимодействии двух бифункциональных мономеров, трудно предположить при данных условиях проведения реакции, исключающих образование биуретов. По всей вероятности, наблюдаемые эффекты можно объяснить тем, что после первых актов реакции существенную роль начинают играть специфические взаимодействия типа водородных связей, которые связывают уретановые и карбамидные фрагменты цепей, склонные к образованию водородных связей [8, 9], и концевые изоцианатные группы.
Такое положение приводит к некоторому занижению концентрации изоцианатных групп, определяемых при используемом методе химического анализа, поскольку кажущееся уменьшение функциональных групп превышает и то, которое расходуется с химической реакцией. Вследствие этого наблюдается перегиб на графике зависимости степени поликонденсации от времени (рис. 1, б) и соответственно отклонение зависимости от расчетной при величинах констант, определенных для начального участка реакции (рис. 2). Это в конечном счете, видимо, приводит к появлению разветвлений с узлами сетки, образованными специфическими взаимодействиями, и явлениям, подобным наблюдаемым при образовании сетки химических связей.
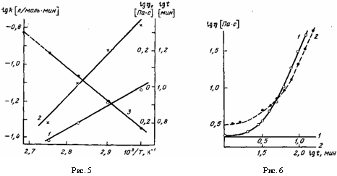
Рис. 5. Зависимость времени достижения уровня вязкости 1,4 Пас (1), а также начальной вязкости реакционной массы (2) и константы скорости поликонденсации (3) от температуры
Рис. 6. Изменение вязкости в процессе поликонденсации ПУ: 1 - бифункциональный отвердитель (3,3'-дихлор-4,4'-диаминодифенилметан), 2 - трехфункциональный отвердитель (по данным работы [7])
Литература
1.Синтез и физикохимия полиуретанов. Киев: Наукова думка, 1967.
2.Mussati F.G., Macosko С.W. Polymer Engng Sci., 1973, v. 13, N 3, p. 236.
3.Kamal M.R., Polymer Engng Sci., 1974, v. 14, N 4, p. 231.
4.Райт П., Камминг А. Полиуретановые эластомеры. Л.: Химия, 1973, с. 303.
5.Малкин А.Я. Успехи химии, 1981, т. 50, № 1, с. 137.
6.Куличихин С.Г., Кожина В.А., Болотина Л.М., Малкин А.Я. Высокомолек. соед. Б, 1982, т. 24, № 4, с. 309.
7.Lipshitz S.D., Macosko С.W. Polymer Engng Sci., 1976, v. 16, N 12, p. 803.
8.Вулканизация эластомеров / Под ред. Аллигера Г., Сьетуна И.М.: Химия, 1967, с. 355.
9.Сергеева Л.М., Липатов Ю.С, Бинъкевич Н.И. В кн.: Синтез и физикохимия полиуретанов. Киев: Наукова думка, 1967, с. 131.
0 комментариев