Федеральное агентство по образованию
Московская государственная академия
тонкой химической технологии
им. М.В. Ломоносова
Факультет: Биотехнологии и органического синтеза
Специальность: 250700 «Технология кинофотоматериалов и магнитных носителей»
Кафедра: Химии и технологии биологически активных
соединений им. Н. А. Преображенского
Квалификационная работа специалиста
Тема.
Москва 2006 г.
Пятичленные гетероциклические структуры, такие как пиррол, а также сопряженные молекулы, содержащие пиррольные фрагменты потенциально важны в качестве оптических электроактивных материалов. Два класса таких материалов активно изучаются: фотолюминесцентные гетероциклические соединения, как призводные бензохинолина, бензоксазола, оксодиазола и фталоцианина, которые проявляют высокую фотолюминесцентную активность в растворах [1,2]; и фотопроводники и электролюминесцентные соединения для электролюминесцентных устройств, большинство из которых являются гетероциклическими соединениями [3]. Присутствие гетероатома в молекуле обеспечивает инжектирование и транспорт электронов или дырок, что необходимо при создании материалов с электроактивными слоями. Пиррольная структура является примером таких молекул. Если молекула содержит два связанных кольца, то наблюдается высокая степень планарности и в результате приводит к улучшению оптических и электронных свойств.
Хорошо известны и изучены дикетопирролпиррольные структуры (DPP), которые являются примером сопряженных молекул с двумя связанными кольцами[4]. Они нашли применение в качестве красного пигмента в промышленности, в частности составляющей красной автомобильной краски[5], газового сенсора для определения водорода [6], флюоресцентных индикаторов для определения концентрации внутриклеточного Ca2+ [7].
Сопряженные пирролы также являются исходными соединениями в синтезе высоко сопряженных порфиринов. Порфириновые ядра, содержащие дополнительные гетероароматические или гетероциклические фрагменты поглощают в более длинноволновой области спектра, чем незамещенные порфирины[8]. Возможны два подхода к синтезу конденсированных пирролов и, в частности, пирролотиазолов – образование на основе пиррола тиазольного гетероцикла и другой – построение пиррольного гетероцикла на основе тиазола. Последний подход широко используется для получения расширенных пирролов, интермедиата в синтезе порфиринов. В новой работе Лаша [9] расширенные пирролы получали реакцией Бартона-Зарда. Японские химики опубликовали работу, где описан синтез пирролобензотиазолов [10]. Попытка синтезировать тиазолопирролы по Бартону-Зарду была неудачна [11].
Таким образом, конденсированные системы на основе пиррола являются потенциально важными соединениями, однако, методы их синтеза плохо разработаны и актуальной задачей является разработка новых подходов к их синтезу.
Для синтеза пиррольных интермедиатов существует большое количество методов широко представленных в обзорах [12]. В данной работе мы рассмотрим некоторые классические и новые методы синтеза замещенных пирролов, а также их реакции.
Классические методы синтеза замещенных пирролов
Синтез Кнорра наиболее общий и широко используемый метод получения пирролов, где происходит образование связей C-N и C-C в результате реакции аминогруппы и метиленовой группы с карбонильной [13]. Он заключается в конденсации a-аминокетонов и a-амино-b-кетоэфиров с кетонами или кетоэфирами в присутствии уксусной кислоты и реже щелочи. Реакции обычно протекают с хорошим выходом. a-Аминокетоны получают восстановлением цинком в уксусной кислоте из предварительно полученных изонитрозо-b-кетоэфиров или изонитрозо-b-дикетонов[14,15].
Не менее интересной и важной является реакция взаимодействия 1,4-дикарбонильных соединений с аммиаком по Паалю-Кнорру [16,17]. Это конденсация, при которой в готовый углеродный скелет вводится атом азота при помощи аммиака или аминов. Механизм реакции, очевидно, включает нуклеофильное присоединение аммиака к двум карбонильным атомам углерода и последующее отщепление воды [18].
Отмечено также, что реакция может протекать с ацетатом аммония с хорошим выходом (~70%), причем, чем более электроноакцепторные заместители в 1,4-дикетоне, тем в более жестких условиях протекает реакция [19].
К этой же группе реакций можно отнести получение пирролов по Ганчу из a-галогенкетонов, b-кетоэфиров и аммиака [20].
Согласно предложенному механизму, сначала происходит образование С-С связи и возникает g-дикетон, который далее реагирует с амином. К реакциям этой группы относится также взаимодействие аммиака и аминов с полиокси- и полигалоидными соединениями [21].
Новые методы синтеза замещенных пирролов
Новой модификацией метода Пааля-Кнорра является синтез замещенных пирролов (2) с использованием нитрата висмута, как катализатора. Это реакция 2,5-дикетонов(1) и основных ароматических аминов в присутствии 5% раствора нитрата висмута в дихлорметане. Мягкие условия и высокие выходы (~80%) продуктов отличительная особенность данного синтеза.
Для доказательства важности нитрата висмута в роли катализатора использовали и другие его соли. Однако положительных результатов не было получено. С разными выходами реакция может протекать и при замене 2,5-дикетона на ди- или монозамещенные дикетоны [22].
В реакции Клауса-Касса замещенные пирролы получают мягким гидролизом 2,5-диметокситетрагидрофурана (3), в результате чего образуется 2,5-дигидрокситетрагирофуран, который в ацетатном буферном растворе при комнатной температуре реагирует с первичными аминами дает N-замещенные пирролы (4) с высокими выходами и чистотой (~89-94%). При проведении реакции в жестких условиях, то есть при высоких температурах и сильнокислотных условиях, происходит разрушение структуры пиррола [23].
Другим возможным вариантом получения замещенных пирролов (7) с использованием катализатора стала реакция алкинов, содержащих легкоуходящие группы (EWG1) (5) c изоцианидами, имеющими объемные заместители (EWG2) (6). Катализатором служат фосфорорганические соединения. Протекают реакции с хорошим выходом (~60%) .
R=Me, Ph, t-Bu, CO2Et.
EWG1=CO2Et, CN.
EWG2= CO2Bu, CONEt2, P(O)(OEt)2
Dppp-1,3-бис(дифенилфосфино)пропан.
Предложенный механизм реакции предполагает нуклеофильное присоединение фосфорорганического катализатора к замещенному алкину с образованием промежуточного продукта. От изоцианида отрывается кислый протон с образованием карбаниона, который атакует атом углерода интермедиата со стороны EWG1 группы и образуется новый анионный центр. Далее происходит [3+2] циклоприсоединение и в итоге получается конечный продукт пиррол [24].
Данный способ нашел применение для синтеза муравьиного ферромона.
Также замещенные пирролы можно получить при взаимодействии гомохиральных первичных аминов (9), аминоспиртов и a-аминоэфиров с 2-пропенил-1,3-дикарбонильными соединениями (8) на золотом катализаторе. Выходы полученных продуктов, а именно 1,2,5-тризамещенных-3-ацилпирролов (10) очень высоки (~95%)
Реакция первичных аминов с 2-пропенил-1,3-дикарбонилом дает производные енамина, который подвергается региоселективному циклоаминированию в пиррол под действием NaAuCl4*5H2O катализатора. Предположительно это происходит путем антиприсоединения атома азота и частично золота по 5 положению, образуя ацетиленовую связь, винилауратного типа. Последующие протолиз связи Csp2---Au и реакция изомеризации дают замещенные пирролы [25].
Использование мягких реакционных условий при проведении реакции с 2-пропенил-1,3-дикарбонильными соединениями позволяет избежать рацемизации.
Регио- и хемоселективность взаимодействия с ацетиленовыми связями одна из интересных особенностей катализатора на основе Au (III). Несмотря на различные металлические соли, успешно катализирующие реакции внутримолекулярного присоединения аминов к кетонам, ²золотой² катализатор, как показано, обладает большей активностью в таких конденсациях.
Система, включающая TiCl4 и t-BuNH2, действует как катализатор для региоселективных реакций гидроаминирования алкинов. Гидразины в этих условиях дают гидразоны, перегруппировывающиеся в производные индола (~76%).
Реакции гидроаминирования несимметрично замещенных алкинов происходят с высокой региоселективностью.
Пирролы (11) получаются при реакции производных анилина и 1,3-диинов под действием TiCl4 и t-BuNH2 при 105º (~30%), в результате аминирования тройных связей [26].
Циклизация α-аминоалленов (12), катализируемая палладием позволяет получить пирролы (13). Реакция протекает с высоким выходом (~55%). Большое значение в этом методе придается условиям реакции, потому что также могут получаться пирролины [27].
Мартин Рейсер и Герхард Маас предложили следующий способ получения пирролов из енаминкетонов (14) [28]. 1-Диалкиламино-1,3-диарил-3-дифенилфосфанилаллены (15), как промежуточные соединения, термически превращаются в 3,5-диарилпирролы (16). Эти превращения, вероятно, заключаются в том, что сопряженные азометиновые илидные интермедиаты подвергаются или 1,5- или 1,7-циклизации. Реакция происходит в три или четыре шага, таким образом, обеспечивается простой синтеза 3,5-диарилпирролов из енаминкетонов. Выход продукта составляет ~60%.
Общий и региоселективный синтез замещенных пирролов (18) путем циклоизомеризации легко осуществить из (Z)-(2-ен-4-винил)аминов (17) (~65%). Происходит произвольная циклоизомеризация и далее присоединение к тройной связи, после чего енамины становятся более стабильными и изомеризуются в соответствующие пирролы при действии металического катализатора[29]. CuCl2 - лучший катализатор для реагентов этой реакции, замещенных по третичному атому углерода. Использование в качестве катализатора производных палладия PdX2 c KX (X = Cl, I) оказалось не эффективным.
В следующей работе [30] описано получение 2,3,4,5-тетра и 2,3,5-тризамещенных пирролов (20). Данный синтез включает в себя три этапа. Исходным соединением является дитиокарбоксилат, который на первом шаге при взаимодействии с этилглицинатом в присутствии триэтиламина дает тиоамид. На втором этапе в результате реакции алкилирования тиоамида образуется кето-N,S-ацеталь (19). Заключительным и самым важным шагом является внутримолекулярная циклизация кето-N,S-ацеталя при действии реагента Вильсмеера-Хака (РОСl3+ДМФА) с образованием замещенного пиррола.
Предложен новый подход для синтеза пирролов [31], который основывается на окислительных свободно-радикальных реакциях производных β-аминокоричной кислоты (21). В этом случае при окислении енаминов церий (IV) тетра-n-бутиламмония нитратом (TBACN) образуются иминные радикалы, которые присоединяются к двойной С–С связи исходного соединения, давая замещенные пирролы (22) с высоким выходом (~87%).
При взаимодействии карбонильного соединения с амином и нитроалкеном в расплаве аммонийной соли получали алкилзамещенные пирролы (23) (~56%). Ни катализаторы, ни органические растворители для этой реакции не требовались.[32]
Реакция енаминов олова (IV) (24) и α-галоальдегидов дает 2,4-дизамещенные пирролы (25) с высоким выходом (~75%) при комнатной температуре, даже в водных условиях [33]. Если проводить реакцию с 2-бромоацетофеноном, то в результате образуются 3,4-дизамещенные пирролы (~64%).
Был осуществлен синтез некоторых новых пирроло[3,4-b]пирролов (28) путем внутримолекулярного циклоприсоединения алкениламиноальдегидов (27) с разнообразными вторичными аминокислотами [34]. Интересно, что во всех случаях происходило образование цис продукта. Конденсация проходила в условиях реакции Дина-Старка в толуоле с высокими выходами (~78%).
Такая же реакция была проведена с N-арилглицинами. В итоге были получены цис продукты с высоким выходом (~75%).
Полифункциональные пирролы (30) можно получить в реакции N-ацетилглицина (29) с реагентами Вильсмеера (ДМФА+ POCl3) с выходом (~89-97%) [35].
Реакция 3,4-диацетил-3-гексен-2,5-диона (31) с алкил или арил первичными аминами дает замещенный пиррол (32) с хорошим выходом (~67%) [36].
При взаимодействии алкилизоцианидов (34) и бензилиден-1,3-дикетонов (33) в результате циклоприсоединения образуется замещенный пиррол (~45%) [37].
Реакции замещенных пирролов
Пиррол относится к электроноизбыточным гетероциклам. Молекула его планарная и ароматичная, а атом азота выступает донором электронов и подает свои электроны в систему, вызывая тем самым увеличение электронной плотности на всем ароматическом кольце пиррола. Реакции обычно проходят по α-положениям, что связано с устойчивостью, образующегося σ-комплекса. При занятых положениях реакция возможна и по β-положениям. Реакции пирролов хорошо известны и подробно описаны в литературе [12]. В данной части литературного обзора приведены новые синтезы, изучение которых проводилось в последнее время.
Реакции электрофильного замещения наиболее характерны для пирролов и большинства его простых производных.
6-Метил-5,6-дигидроиндолизин (35) и 2- или 3-этилпроизводные были получены реакцией электрофильного ароматического замещения из 1-(2-метил-2-пропенил) пирролов. Данный синтез проходит в три шага – гидроформилирование, циклизация, дегидратация. Происходит замещение атома углерода карбонильной группы в α-положение пиррола с образованием шестичленного кольца, что является ключевым моментом в этом процессе.
Реакция проходит в мягких условиях, даже без присутствия кислот Льюиса и выход составляет 53% [38].
Также может происходить присоединение пиррола (36) с N-Tos имином в присутствии Cu(OTf)2 давая пирролосульфамиды (37) с высоким выходом (~70-85%). Присоединение происходит региоселективно по второму положению пиррольного кольца. Данная реакция проста в проведении и не требует безводных условий [39].
Обработка N-алкил-N-аллил-пирроло-2-карбоксамидов (38) каталитическим количеством производных соединений палладия дает региоселективную внутримолекулярную циклизацию с образованием бициклических пиррольных структур. Наиболее вероятна реакция по 1- и 3-положению пиррольного кольца.
Реакция начиналась с окисления и далее с циклизацией по третьему атому углерода пиррольного кольца. В результате получались две изомерные пирролопиридиновые структуры с разными выходами (30 и 35%), которые были выделены. В роли катализатора использовали производные соединения Pd (II). Были проведены реакции с различными субстратами алкил-аллиламинов [40].
Исходный для вышеописанной реакции N-алкил-N-аллил-пирроло-2-карбоксамид (38) может быть легко получен из α-карбоксипиррола (39) [41].2-Формилпиррол (40) может быть пронитрован ацетилнитратом при -40˚С, давая 4- и 5-нитросоединения с общим выходом 71% [42].
Синтез N,N'-дизамещенных дикетопирролопирролов (DPP) (41) проводится в три этапа. На первом происходит взаимодействие этил-2-арил-4,5-дигидро-5-оксопиррол-3-карбоксилата со сложными эфирами или ангидридами в присутствии сильного основания, давая 4-ацил производные, существующие в виде E- или Z-енолов. Следующий шаг заключается в циклизации полученных соединений в растворе при температуре выше 200˚ с образованием 3,6-дизамещенных 1Н-фуро[3,4-с]пирролодионов, которые на заключительном этапе после защиты атома азота пиррольного кольца, реагируют с первичными аминами превращаясь в производные дикетопирролопирролов. Выход конечного продукта составляет 73% [4]
В эту же реакцию могут вступать и неароматические нитрилы, давая новые циклопента[с]пиррол производные (42) (69%). Полученные продукты имеют насыщенную окраску и используются как пигменты. Схема механизма реакции выглядит следующим образом [5].
Реакции нуклеофильного замещения пирролов мало изучены для пирролов, но не менее интересны для исследователя, чем другие типы реакций.
При взаимодействии 3-карбодитиопирролов (43) с СН-кислотами (цианоацетамидом, цианоацетатом) в системе KOH-ДМСО происходит образование функциональных 3-винилпирролов (44), таких как 3(1-алкилтио-2-циано-2-Х-этенил)пиррол (Х =CN, CONH2, CO2Et), при этом важным моментом является то, что замещение происходит не по пиррольному кольцу, положения которого заняты, а по функциональной группе заместителя с выходом 28-58% [43].
Предложен новый метод [44] алкилирования пирролов с такими соединениями, как аллилбромид, кротилбромид в присутствии металлического цинка в тетрагидрофуране, в результате которого получаются соответствующие 3- и 2-алкилпирролы (45), и его производные с хорошим выходом (~56-70%).
Зайцев и соавторы в своей работе [35] изучали региоселективность полифункциональных пирролов в реакциях с нуклеофилами. Исходным соединением для исследования являлся 3,5-дихлоро-1Н-пиррол-2,4-дикарбальдегид (46), который вступает в конденсацию с вторичными аминами, давая метилензамещенные пирролы, а его N-алкил производные путем нуклеофильного замещения по пятому положению дают 5-замещенные пиролы.
Выбранный замещенный пиррол имеет несколько электрофильных центров, таким образом, он может давать ряд продуктов в реакции с нуклеофилами. Особый интерес представляют различия в реакционной способности альдегидных групп во втором и четвертом положениях, что объясняется образованием водородной связи между альдегидной группой и атомом водорода при азоте. Не менее интересны и различие в электрофильности третьего и пятого положений, которое оговаривается действием индуктивного эффекта атома азота на пятое положение, а атом углерода ведет себя как β-углерод в енамине.
Реакция исходного пиррола с морфолином или пиперидином в этаноле при комнатной температуре дает чистые кристаллические продукты, такие как 3,5-дихлоро-2-(1’-пиперидинилметилен)-2Н-пиррол-4-карбальдегид (48) и 3,5-дихлоро-2-(1’-морфолинилметилен)-2Н-пиррол-4-карбальдегид (47) с хорошим выходом (~44-62%).
X = CH2 (62%)
X = O (44%) (48)
Присутствие слабокислого протона N-H группы в пирроле не позволяет провести нуклеофильное замещение Cl группы вторичными аминами, если прежде не проалкилировать пиррол в сухом ДМФА, используя метилйодид или этилбромид как среду, что в результате дает новые производные (49) с высоким выходом (~89-97%).
R = Me, Et, CH2C6H4NO2-2
N-метил и N-этил производные (49) выбрали для дальнейшего исследования реакций нуклеофильного замещения с тиоэтанолом, пиперидином, пирролидином, морфолином и диэтиламином. Реакции с вышеперечисленными реагентами дали продукты замещения по пятому положению в пирроле (~43-86%).
R = Me, Et, CH2C6H4NO2-2
X = EtS, NEt2, 1-пиперидинил, 1-пирролидинил, 1-морфолинил
В реакциях пиррола (49) с морфолином в более жестких условиях (78˚ и 70 часов) происходит образование дизамещенных производных (50) с выходом 21%.
Для получения расширенных порфириновых систем важнейшими интермедиатами являются продукты окисления пирролов по α-положениям.
В литературе описаны способы получения диформилпирролов с помощью церийаммоний нитрата [45] и смеси тетраацетата свинца с диоксидом свинца [46].
В работе Г. Вассермана пирролы подвергаются окислению синглетным кислородом [47]. Трет-бутиловый эфир 3-метокси-2-пирролокарбоксильной кислоты окисляется синглетным кислородом, образуя промежуточный продукт – имино гидропероксид, который может вступать в реакции с различными нуклеофилами давая 5-замещенные пирролы (~56%). Имея сильные нуклеофилы по пятому положению, пиррол может присоединять мало реакционноспособный гидропероксид в результате чего получаются бипиррольные продукты (51).
Сопряженные системы на основе пиррола, например дикетопирролпирролы (DPP) нашли широкое применение в различных областях науки и промышленности (Лит. обзор стр. 22).
Возможны два подхода к синтезу конденсированных пиррольных соединений, а именно к получению пирролотиазолов. В основе первого подхода лежит образование на основе пиррола тиазольного гетероцикла, а второго – построение пиррольного гетероцикла на основе тиазолольного кольца. Последний подход широко используется для получения расширенных пирролов, интермедиатов в синтезе расширенных порфиринов. В одной из последних работ Лаша расширенные пирролы получали реакцией Бартона-Зарда [9]. Мирашима и соавторы разработали синтез пирролобензотиадиазолов реакцией изоцианацетата и нитробензотиадиазола в присутствии сильного основания [10].
Ранее в нашей лаборатории предпринимались попытки получить пирролотиазольные производные конденсацией 2-метил-6-нитробензола и изоцианацетата в присутствии сильных ненуклеофильных оснований, то есть в условиях реакции Бартона-Зарда [11]. Однако данный подход не дал положительных результатов, вероятно из-за повышенной кислотности протона в тиазольном кольце, которая мешает отщеплению протона нитробензольного кольца, что является необходимым условием для протекания данной реакции.
Другой подход заключается в получении тиазольного цикла на основе замещенного пиррола. В литературе не описано построения гетероциклического фрагмента на пиррольном цикле. В итоге мы решили воспользоваться условиями реакцией Ганча [20], как образцом, с помощью которой успешно синтезируют тиазолы взаимодействием α-галогенкетонов и тиоациламидов. Важно учитывать, что в данном синтезе могут использоваться в качестве исходных соединений только гидроксигалоген- или дигалогензамещенные пирролы.
Таким образом, ключевое вещество в нашей схеме - 2,5-диметил-3,4-дийодпиррол. Схема синтеза данного соединения была тщательно разработана.
На первом этапе исследования был получен 2,5-диметил-3-этоксикарбонилпиррол по следующей схеме:
На первой стадии нарабатывался в достаточном количестве монобромацетон (1), который получали по известной методике [48] взаимодействием ацетона с бромом в водной уксусной кислоте при температуре 70˚С в течение 30 минут.
Бромирование в кислой среде позволило избежать получения ди- и полибромпроизводных ацетона и получить монобромацетон с выходом 40%.
Следующая стадия синтеза – получение ацетонилацетоуксусного эфира (2). Реакцию алкилирования проводили двумя способами, отличающимися использованием в реакции алкилирования различных растворителей.
Первый способ заключался в проведении реакции алкилирования в этаноле. То есть первоначально в этанольном растворе получали этилат натрия, который и служил основанием при синтезе натрацетоуксусного эфира. Реакцию проводили в при температуре 40-60˚С в течение 30 минут. Выход продукта составил 55%.
Во втором случае натрацетоуксусный эфир получался непосредственно при действии металлического натрия на ацетоуксусный эфир в растворе диэтилового эфира. То есть из схемы процесса убиралась стадия получения этилата натрия. Выход продукта по этой схеме составил 60-65%. Упрощение схемы получения продукта, сопровождающееся некоторым повышением выхода целевого соединения делает второй способ получения ацетонилацетоуксусного эфира (2) предпочтительным.
Для синтеза 2,5-диметил-3-этоксикарбонилпиррола (3) была выбрана конденсация производных 1,4-дикарбонильных соединений с ацетатом аммония, выступающего в роли поставщика аминного фрагмента, то есть по реакции Пааля-Кнорра [49]:
Выход в реакции составил 83%. Константы выделенного продукта совпали с приведенными в литературе [50]. В ИК-спектре присутствуют характерные полосы колебаний N-H связи – 3300см-1, C=O связи – 1659см-1, С=С связи – 1600см-1 и C-O связи – 1220см-1 и 1090см-1.
Следующим этапом было получение целевого 2,5-диметил-3,4-дийодпиррола (5) по следующей схеме:
Первая стадия - это омыление 2,5-диметил-3-этоксикарбонилпиррола (3), которое осуществлялось кипячением (60-70ºC) с 30% NaOH в водном метаноле в течение 4 часов. Далее реакционную массу нейтрализовали концентрированной соляной кислотой при температуре не выше 10ºС для избежания декарбоксилирования пирролкарбоновой кислоты. Выпавший в результате нейтрализации осадок отфильтровывали и сушили на воздухе. Так как в выпавшем осадке наряду с карбоновой кислотой может присутствовать NaCl, то для выделения органического продукта была проведена его экстракция ацетоном. После упаривания ацетона выход 2,5-диметил-3-карбоксипиррола (4) составил 63%. Соединение было охарактеризовано ИК- и ПМР-спектроскопии. В ИК-спектре присутствовали характерные полосы колебаний N-H связи – 3260см-1, ОН при 3000-2500 см-1 и С=О при 1639 см-1. ПМР (D2O) δ (м.д.): 1,92-с. (3Н; CH3); 2,21-c. (3H; CH3); 5,81-c. (1H;CH).
Полученный 2,5-диметил-3-карбоксипиррол (4) иодировали в водно-метанольном растворе в присутствии поташа раствором йода и KI в воде при температуре 65˚С, далее реакционную массу перемешивали в течение 20 минут и охлаждали. Выпавший осадок отфильтровывали и промывали большим количеством воды. Выход 2,5-диметил-3,4-дийодпиррола (5) составил 40%. Соединение было охарактеризовано физико-химическими и спектральными методами и имеет следующие характеристики: Rf = 0,5 (ПЭ:ЭА 1:1); ПМР δ (м.д.): 2,28-с. (6Н; СН3), 8,01-уш. с. (1Н; NH); Масс спектр m/z (%): 347 (100%).
Полученный 2,5-диметил-3,4-дийодпиррол (5) мы использовали для изучения нуклеофильных реакций, что и являлось целью нашей работы. Как отмечалось в литературном обзоре (стр. 19), нуклеофильные реакции для пиррольных соединений не характерны и мало изучены, поэтому нам предстояло найти растворители, в которых эти реакции протекают с наибольшими выходами и установить температурные режимы их проведения. В качестве нуклеофильных реагентов нами были взяты для исследования следующие соединения - нитрит серебра, тиоацетамид, тиомочевина, ортофенилендиамин. Выбор нитрита серебра был обусловлен тем, что ранее мы показали [51], что нитрит серебра эффективно и с высоким выходом замещает галоген в β-положении на нитрогруппу. Необходимый для этого исследования 3-йод-2,5-диметил-4-этоксикабонилпиррол (6) получали из 2,5-диметил-3-этоксикарбонилпиррола (3) в водном метаноле действием йода в присутствии KI и поташа. Выпавший осадок отфильтровывали и сушили под вакуумом. Выход 3-йод-2,5-диметил-4-этоксикабонилпиррола (6) составил 80%.
Для получения максимального выхода 2,5-диметил-3-нитро-4-этоксикарбонилпиррола (7) оптимизировали условия проведения реакции. Во-первых, был осуществлен подбор растворителя. Были изучены следующие растворители – диэтиловый эфир, ацетонитрил и ДМФА. В диэтиловом эфире реакция вовсе не начиналась. Низкие выходы (20%) получались и при использовании ДМФА+мочевина (мочевина добавляется для увеличения растворимости исходного реагента), что объясняется сложностью выделения полученного продукта, так как реакционную смесь обрабатывали водой, чтобы избавиться от ДМФА, а продукт, хорошо растворяющийся в воде, трудно затем из нее извлекался. В итоге, оптимальным растворителем оказался ацетонитрил. Во-вторых, изучали влияние температуры на ход процесса. Попытки проведения реакции при 0º и при комнатной температуре не принесли результата, тогда как при кипячении реакция проходит. Оптимальным временем проведения реакции оказалось 2 часа при кипячении, так как дальнейшее увеличение времени реакции не улучшило результат. Таким образом, наибольшие выходы (47%) в этой реакции удалось получить при кипячении 3-йод-2,5-диметил-4-этоксикабонилпиррола (6) с нитритом серебра в ацетонитриле в течение 2 часов в темноте, так как соли серебра под действием света разлагаются. Далее реакционную массу отфильтровывали от соли серебра и продукт очищали с помощью колоночной хроматографии на силикагеле в системе растворителей петролейный эфир:этилацетат (1:1). Полученное вещество было охарактеризовано с помощью ИК-, ПМР-спектроскопии и масс-спектрометрии. В ИК-спектре продукта помимо полос N-H (3310 см-1), С=О (1680 см-1) и С-О (1020 см-1 и 1110 см-1) присутствующих в исходном соединении появились интенсивные полосы, характерные для колебаний N=О (1680 и 1600 см-1) и C-N (1530 см-1) групп. В ПМР произошло смещение сигналов протонов метильных групп с 2,15 до 2,35-с. (3Н, СН3) и с 2,45 до 2,48-с. (3Н, СН3). В масс-спектре наблюдались пик молекулярного иона m/z = 212 (19%), а также пики устойчивых фрагментов: m/z = 166 (40%), 122 (43%), 92 (45%), 42 (100%).
Для проведения реакции с 2,5-диметил-3,4-дийодпирролом (5) мы выбрали те же реакционные условия, то есть реакцию замещения йода на нитрогруппу проводили при кипячении 3,4-дийодпиррола (5) с нитритом серебра в ацетонитриле в течение 2 ч или при выдерживании той же смеси в течение суток. Из реакционной смеси отфильтровывали осадок солей серебра и экстрагировали из него продукт ацетоном. По данным ТСХ реакционная масса состояла в основном из индивидуального продукта (Rf = 0,8(ПЭ:ЭА 1:1)), но содержала небольшую примесь исходного 3,4-дийодпиррола (Rf = 0,5(ПЭ:ЭА)) и незначительное количество других примесей. Однако, выделенный основной продукт оказался не продуктом замещения. Молекуярная масса полученного соединения по данным масс-спектрометрии составляла 361. В ИК-спектре помимо полос характерных для пиррольного гетероцикла, наблюдалась интенсивная полоса колебаний связи С=О при 1670 см-1. Характерных для нитро-группы сигналов N=О (1680 и 1600 см-1) и C-N (1530 см-1) групп не наблюдалось. В спектре ЯМР 1Н появился новый синглетный сигнал протона при 9.35 м.д. Совокупность полученных данных позволила заключить, что основным процессом было окисление одной из метильных групп до формильной. Вероятно, окислителем послужила соль серебра. Продукт окисления – 3,4-дийод-2-метил-5-формилпиррол (8) был выделен в виде кристаллического вещества, достаточно быстро разлагающегося на воздухе, что не позволило определить его температуру плавления. Выход продукта после очистки составил 11%.
Выбор следующих двух реагентов, был обусловлен нашим желанием получить конденсированные пирролы, смоделировав условия реакции Ганча [20]. Первым реагентом, с которым мы проводили исследования, был тиоацетамид. Синтез проводили двумя разными способами:
В первом случае растирали оба компонента, без растворителя при комнатной температуре и оставляли стоять в течение 10-15 мин. Наблюдали почернение реакционной массы, сопровождаемое повышением температуры смеси и выделением резкого неприятного запаха. Продукт отмывали бензолом и очищали с помощью колоночной хроматографии на силикагеле в системе растворителей гексан : этилацетат (8:1). В результате получили 2-метилпирроло[3,4-d][1,3]тиазол (9) с выходом 32%. Полученный продукт представляет собой кристаллическое вещество, и его структура была подтверждена спектральными методами. На ПМР спектре присутствуют синглетные сигналы протонов метильных групп во 2 и 5 положениях пиррольного кольца, которые смещены по сравнению с исходным соединением в слабое поле:
-СН3: 2,42 м.д. (3Н, с),
-СН3: 2,48 м.д. (3Н, с).
Помимо этих сигналов, присутствует сигнал протонов метильной группы –СН3 тиазольного кольца 2,58 м.д. (3Н; с), а также протона N-H группы: 7,94 м.д. (1Н; уш.с.). В масс-спектре соединения наблюдается молекулярный ион (m/z+1) 167.
Для повышения выхода продукта нами была проведена оптимизация условий получения. Во-первых, был осуществлен подбор растворителя. Оптимальным для этого синтеза оказался метанол. При попытке проведения реакции в других растворителях, таких как ДМФА и ацетонитрил, реакция даже не начиналась. Выбор метанола, как растворителя, кроме всего, определялся тем, что оба компонента реакции в нем хорошо растворяются, и он используется в условиях метода Ганча [20]. Во вторых, изучили влияние температуры и времени на ход процесса. Для начала реакционную массу грели при температуре 30ºС в течение 5 мин, однако по данным ТСХ не обнаружили увеличения пятна продукта реакции и уменьшения пятен исходного вещества и побочных продуктов. Далее постепенно повышали температуру на 10ºС и варьировали временной интервал от 5 до 30 мин. В итоге, по данным ТСХ обнаружили, что при проведении реакции в течение 5 мин и температуре 40ºС исчезает пятно исходного реагента, а пятно продукта увеличивается. Дальнейшее повышение температуры и изменение временного интервала не дало положительного результата. Поэтому для получения максимального выхода продукта первоначально оба вещества по отдельности растворяли в метаноле, а затем сливали оба раствора, смесь грели в течение 5 мин при 40º и оставляли стоять при комнатной температуре в течение 10-15, что приводило, как показали данные ТСХ, еще и к уменьшению количества примесных пятен. Продукт очищали с помощью колоночной хроматографии на силикагеле в системе растворителей гексан: этилацетат (3:1). В итоге получили 2-метилпирроло[3,4-d][1,3]тиазол с выходом (9) 56%.
Успешно проведя реакцию замещенного пирролотиазола мы решили дальше использовать синтетические возможности обнаруженной реакции.
Следующей конденсацией была реакция 3,4-дийодпиррола (5) с тиомочевиной. Данную реакцию проводили, взяв за основу выше отработанную методику. При проведении реакции в метаноле в течении 20 мин при комнатной температуре реакция не начиналась. Для увеличения полярности растворителя была взята водно-метанольная смесь (1:1). Для облегчения отрыва галогена добавляли поташ. В итоге, синтез проводили при взаимодействии 2,5-диметил-3,4-дийодпиррола (5) и тиомочевины в присутствии поташа (в эквимолярном соотношении) и в водно-метанольном растворе. По данным ТСХ реакция закончилась через 20 мин выдерживания при комнатной температуре. Продукт отмывали четыреххлористым углеродом от основного количества побочных продуктов и очищали с помощью колоночной хроматографии на силикагеле в системе растворителей ПЭ:ЭА (4:1). Выход продукта составил 43%. 2-аминопирроло[3,4-d][1,3]тиазол (10) представляет собой маслянистое вещество желтого цвета, достаточно быстро разлагающееся на воздухе. Его структура была подтверждена ПМР- и масс-спектроскопией. Помимо синглетных сигналов метильных групп – 2,22 м.д. (6Н; СН3) и сигнала уширенного синглета N-H группы – 7,7 м.д. (1Н; NH), на ПМР-спектре наблюдается дублетный сигнал протонов NH2-группы: 5,87 м.д. (2Н; NH2). На масс-спектре присутствует молекулярный ион m/z 168.
Два предыдущих эксперимента представляют собой конденсации с N,S-нуклеофилами, интересным было исследовать проведение аналогичной конденсации с N,N-нуклеофилом. В качестве последнего был выбран ортофенилендиамин. Реакцию проводили в тех же условиях, что и при получении 2-аминопирроло[3,4-d][1,3]тиазола (10), однако время реакции пришлось увеличить. Пятно 3,4-дийодпиррола (5) на хроматограмме исчезло только через 40 мин. При этом на ТСХ помимо основного пятна наблюдалось большое количество пятен примесей. Ни увеличение времени реакции до 2 часов, ни повышение температуры реакции не позволило уменьшить содержание побочных продуктов, представляющих проблему для дальнейшего выделения продукта. Экстрагирование продукта из реакционной смеси четыреххлористым углеродом помогло избавиться лишь от малой части побочных продуктов. При помощи колоночной хроматографии на силикагеле в системе растворителей ПЭ:ЭА (4:1) удалось выделить индивидуальное маслянистое вещество с выходом 25%, которое оказалось неустойчивым при хранении, даже при пониженной температуре. Анализ ПМР-спектра показал, что произошло присоединение фенильного заместителя к пиррольному кольцу (наблюдаются сигналы четырех протонов в области 7 м.д. в виде двух симметричных групп сигналов, состоящих из триплета (КССВ 7,1 и 6,8 Гц) и синглета). Наличие сигнала в области 5,66 м.д. подтверждает присутствие NH протонов, а два синглета интенсивностью по 3Н в области 2,19 и 2,13 м.д. говорит о присутствии в соединении двух неэквивалентных метильных групп. Указанный характер ПМР-спектра позволяет предположить, что замещение произошло не по обоим β,β'-положениям пиррольного кольца, а только с замещением одной йод группы с образованием несимметричной структуры. Остальные спектральные анализы не удалось провести, так как полученное вещество быстро разлагалось, что приводило к трудности выделения его из общей смеси.
Так как пирролотиазолы могут оказаться интересными люминесцирующими соединениями, было проведено первичное изучение их электронных спектров поглощения и флуоресценции.
Электронные спектры поглощения 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола(9) и 2-аминопирроло[3,4-d][1,3]тиазола(10) характеризуются интенсивной полосой в коротковолновой области спектра с пиком 235 нм и плечом 276 нм для соединения(9) (Рис 1) и пиком 225 нм с плечом 275 нм для соединения(10) (Рис 2). Сравнивая полученные данные со спектром исходного 3,4-дийодпиррол(5), имеющего пик 248нм с плечом 310 нм, можно сказать, что полученные 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола(9) и 2-аминопирроло[3,4-d][1,3]тиазола(10) демонстрируют батохромный эффект.
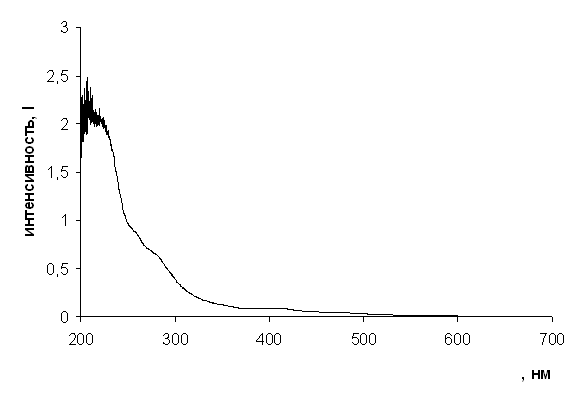
Рис. 1 Спектр поглощения 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола в гексане.

Рис. 2 Спектр поглощения 2-амино-4,6-диметил-5Н-пирроло[3,4-d][1,3]тиазола в гексане.
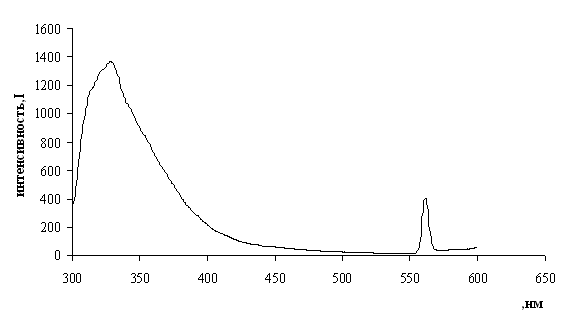
На спектрах флуоресценции 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола(9) и 2-аминопирроло[3,4-d][1,3]тиазола(10) наблюдаются интенсивные пики с максимумами соответственно в 328 нм (Рис 3) и 310 нм (Рис 4), тогда как исходный 3,4-дийодпиррол(5) не флуоресцирует.
Рис. 3 Спектр флуоресценции 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола в гексане.
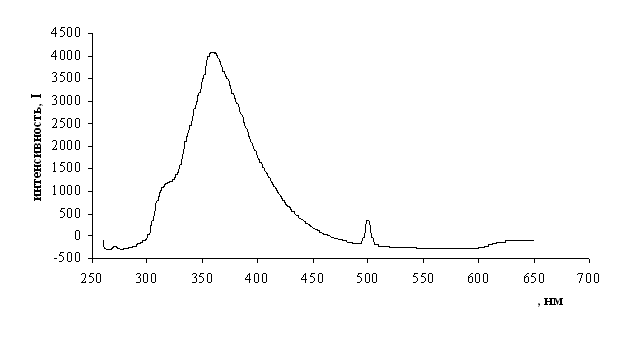
Рис 4. Спектр флуоресценции 2-аминопирроло[3,4-d][1,3]тиазола в гексане.
В данной работе использовали вещества и растворители с характеристиками ХЧ, ЧДА и ацетонитрил с характеристикой ОСЧ.
Для идентификации синтезируемых соединений и контроля протекания реакций использовали ТСХ на пластинах "Silufol UV 254" и "Merck 60 F 254" в различных системах растворителей. Проявляли в парах йода.
Колоночную хроматографию проводили на силикагеле фирмы Merck Silica gel 60 в различных системах растворителей.
Спектры 1H-ЯМР регистрировались на импульсном фурье - спектрофотометре Bruker AMX – 400 (Германия) с рабочей частотой 400 Мгц в CDCl3 (δ м.д.), внешний стандарт тетраметилсилан.
ИК-спектр в вазелиновом масле и тонком слое вещества снимали на спектрофотометре "Shimadzu IR-435"(Япония).
Масс-спектры снимали на масс-спектрометрах "Finnigan MAT INCOS 50", метод-электронный удар 70 эВ и "Kratos PC-Kompact MALDI 4" (США), метод испарение лазером (регистрирующем технологию MALDI-TOF-Matrix Assisted Laser Desorption Ionisation Time-Of-Flight Mass Spectrometry). В качестве матрицы для низко-молекулярных органических соединений использовали гентистик-2,5-дигидроксибензойная кислота.
Электронные спектры поглощения записывали на спектрофотометре Jasco UV 7800(Япония).
Спектры флуоресценции определяли на спектрофлуориметре Jasco FP-777(Япония).
Температуру плавления определяли с помощью прибора для определения температуры плавления "Boetius" (Германия).
1-Бром-2-пропанон.(1)
В трехгорлую колбу на 500 мл с обратным холодильником, мешалкой, термометром и капельной воронкой помещали 190 мл воды, 58,6мл (0,8 моль) ацетона и 44мл (0.76 моль) уксусной кислоты. При температуре 700C прибавляли по каплям 41мл (0,8 моль) брома до исчезновения окраски, затем раствор перемешивали при той же температуре 10 мин, добавляли к нему 100 мл холодной воды и охлаждали до 5-100C. Смесь нейтрализовали твердой содой до рН=6-7 и отделяли органический слой, который сушили над безводным MgSO4. Перегоняли в вакууме. Получили 26.0 мл (40%) 1-бром-2-пропанона(1) с nD25=1,4693 Tкип.=43-470/25-30 мм.
Лит: nD20=1,4697 Ткип.=136,50 [52]
Ацетонилацетоуксусный эфир.(2)
Вариант 1В трехгорлую колбу на 250 мл с обратным холодильником, мешалкой, термометром и капельной воронкой помещали 59 мл абсолютного этанола и прибавляли небольшими порциями 3,5 г (0,15 моль) натрия до растворения и образования этилата натрия, далее при комнатной температуре добавляли по каплям 23,0 мл (0,18 моль) ацетоуксусного эфира. Реакционную массу охлаждали до 200C и прикапывали 13,0 мл (0,15 моль) бромацетона(1) таким образом, чтобы температура не превышала 400С, а затем температуру смеси повышали до 600С и перемешивали в течение 10 мин. Смесь охлаждали до 200С, этанол отгоняли на роторном испарителе и добавляли небольшое количество воды для растворения NaBr. Затем с помощью делительной воронки отделяли нижний водный слой и экстрагировали из него продукт диэтиловым эфиром. Экстракт сушили над безводным MgSO4. Растворитель отгоняли на роторном испарителе, и остаток перегоняли в вакууме. Получили 17,0мл (55%) ацетонилацетоуксусного эфира(2) с nD22=1,4378 T кип.=1440/21мм.
Лит: nD20=1,4375 Т кип.=145-1460/21мм. [ 48]
Вариант 2
В трехгорлую колбу на 500 мл снабженную механической мешалкой, обратным холодильником и термометром помещают 100 мл диэтилового эфира, 28,3 мл (0,22 моль) ацетоуксусного эфира и при 250С добавляют небольшими порциями 14,6 г (0,63 моль) натрия, температура возрастает до 360С. После образования густого осадка приливают 250 мл диэтилового эфира для лучшего перемешивания, далее оставляют на 36 часов. К полученной реакционной массе по каплям прибавляют 17 мл (0,20 моль) бромацетона(1). Перемешивают 2 часа и приливают 30 мл воды до растворения осадка. Отделяют нижний водный слой, экстрагируют диэтиловым эфиром. Объединенные слои сушат над безводным MgSO4. Растворитель отгоняют, остаток перегоняют в вакууме. Получают 18,8 мл (60%) ацетонилацетоуксусного эфира(2) с nD22=1,4377 T кип.=1440/21мм.
Лит: nD20=1,4375 Ткип.=145-1460/21мм.[48]
2,5-Диметил-3-этоксикарбонилпиррол.(3)
В колбе на 200 мл с механической мешалкой смешивали 17,0 мл (0,1 моль) ацетонилацетоуксусного эфира(2), 22,0 г (0,28 моль) ацетата аммония, 50 мл уксусной кислоты и 13,0 мл уксусного ангидрида. Реакционную массу перемешивали 20 мин и прибавляли к ней 100 мл холодной воды. Затем смесь оставляли в холодильнике на сутки в результате выпал осадок, который отфильтровали и оставили сушить под вакуумом. Получили 16,7 г (83%) 2,5-диметил-3-этоксикарбонилпиррола(3) с Tпл.=1130. ПМР d (м.д.):1.30-тр. (3H; -CH2CH3; J=7.24 Гц) 2.17–с. (3H; CH3); 2.45–c. (3H; CH3); 4.23–кв. (2H; CH2; J=7.24 Гц); 6.17–д. (1H; CH, J=2.14 Гц); 8.09‑уш.c. (1H; NH).
Лит: Тпл.=1130. [49]; ПМР (CDCl3) d (м.д.): 1.33-т. (3H; CH3; J=7.2 Гц); 2.19-с. (3H; CH3); 2.47-с. (3H; CH3); 4.25-кв. (2H; CH2; J=7.24 Гц); 6.20-с. (1H; CH); 8.17-уш.с. (1H; NH)[48].
2,5-Диметил-3-карбоксипиррол.(4)
В колбе на 50,0 мл с обратным холодильником кипятили смесь 5,0 г (0.03 моль) 2,5-диметил-3-этоксикарбонилпиррола(3) в 36,0 мл метанола и 18,0 мл 30% водного раствора NaOH в течение 3 часов 20 мин. Затем реакционную массу охлаждают и нейтрализуют концентрированной HCl. Выпавший осадок отфильтровывают и сушат на воздухе. Получили 2,62 г (63%) 2,5-диметил-3-карбоксипиррола(4) с Rf=0,6(ПЭ:ЭА 1:1); Тпл.=1820C. ПМР (D2O) δ (м.д.): 1,92-с. (3Н; CH3); 2,21-c. (3H; CH3); 5,81-c. (1H; CH). ИК (вазелиновое масло) ν (см-1): 3260, 3000-2500, 1639.
2,5-Диметил-3,4-дийодпиррол.(5)
В трехгорлую колбу на 150 мл с дефлегматором, мешалкой, капельной воронкой и термометром вносили 0,5 г (0,0036 моль) 2,5-диметил-3-карбоксипиррола(4) в 20 мл метанола. При перемешивании в полученную суспензию прикапывали раствор 1,24 г (0,009 моль) К2СО3 в 20 мл воды и нагревали до 650С, при этой температуре добавляли раствор 0,76 г (0,003 моль) I2 и 1,15 г(0,007 моль) КI в 10 мл воды. Реакционную массу перемешивали 20 мин. и охлаждали. Выпавший осадок отфильтровали и промывали большим количеством воды. Получили 0,44 г (33%) 2,5-диметил-3,4-дийодпиррола(5) с Rf=0,5(ПЭ:ЭА 1:1); Tпл.=1170С. ПМР δ (м.д.): 2.28-с. (6Н; СН3); 8,01-уш.с. (1Н; NH); Масс спектр m/z (%): 347 (100%).
2,5-Диметил-3-йод-4-этоксикарбонилпиррол. (6)
В трехгорлой колбе с дефлегматором, мешалкой, капельной воронкой и термометром растворяли 0,5 г (0,003 моль) 2,5-диметил-3-этоксикарбонилпиррола(3) в 20 мл метанола. При перемешивании прикапывали раствор 1,24 г (0,009 моль) K2CO3 в 20 мл Н2О, нагревали до 650С и при этой температуре прибавляли раствор 0,76 г (0,003 моль) I2 и 1,15 г (0,007 моль) KI в 10 мл H2O. Реакционную массу перемешивали 20 мин и охлаждали. Осадок отфильтровали и оставили сушиться под вакуумом. Получили 0,6 г (70%) 2,5-диметил-3-йод-4-этоксикарбонилпиррола(6) с Rf =0,8(ПЭ:ЭА 1:1) и Tпл=1170С. ПМР (CDCl3) d(м.д.):1,34-тр. (3H; -CH2CH3; J= 7,24 Гц) 2,22-c. (3H; CH3); 2,47-c. (3H; CH3); 4,26-кв. (2H; CH2; J= 7,24 Гц); 8,18-уш. с. (1H;NH). 13C-ЯМР (CDCl3) d(м.д.): 13,89 (2-CH3); 14,19 (3-CH3); 14,30 (OCH2CH3); 59,58 (C-I); 63,03 (-CH2-); 129,48 (C-5); 129,55 (C-4); 135,58 (C-2); 164,62 (C=О). ИК (вазелиновое масло) ν (см-1): 3256, 1675, 1217, 1099, 1029, 773. Масс спектр m/z (%): 293 (40%), 279 (20%), 264 (42%), 248 (28%), 219 (18%), 127 (12%), 122 (25%), 93 (30%), 67 (35%), 51 (55%), 42 (100%).
2,5-Диметил-3-этоксикарбонил-4-нитропиррол.(7)
В колбу на 15 мл с обратным холодильником поместили раствор 0,4 г (0,002 моль) 2,5-диметил-3-йод-4-этоксикарбонилпиррола(6) в 5 мл ацетонитрила и добавили раствор 0,42 г (0,003 моль) AgNO2 в 3 мл ацетонитрила. Реакционную массу кипятили 2 ч при Tбани=1100С и оставили на 2 суток при комнатной температуре в темноте. Осадок отфильтровали, продукт очищали с помощью колоночной хроматографии на силикагеле (l=13 см, d=1,6 см) в системе растворителей петролейный эфир: этилацетат (2:1). Получили 0,1 г (47%) 2,5-диметил-3-этоксикарбонил-4-нитропиррола(7) с Rf=0,3(ПЭ: ЭА 1:1) и Тпл.=99-1000C. ПМР (CDCl3) d(м.д.):1,35 м.д.-тр. (3H; CH2CH3, J= 7,1 Гц); 2,35 м.д.-с. (3H; CH3 ); 2,48 м.д.-с. (3H; CH3 ); 4,32 м.д.-кв. (2H; CH2CH3, J=7,1 Гц); 8,91 м.д.-уш.с. (1H; NH). ИК (вазелиновое масло) ν(см-1): 3300, 1720, 1680, 1600. Масс-спектр m/z(%):212 (19%), 166 (40%), 122 (43%), 92 (45%), 66 (28%), 54 (19%), 42 (100%).
3,4-Дийод-2-метил-5-формилпиррол (8).
В колбе с обратным холодильником и мешалкой смешивают 0,9 г (0,0026 моль) 2,5-диметил-3,4-дийод-пиррол(5) в 11,20 мл ацетонитрила и добавляют к нему раствор 0,94 г (0,009 моль) AgNO2 в 6,70 мл ацетонитрила. Реакционную массу кипятят в течение 2 часов, далее охлаждают и оставляют на 2 суток. Выпавший осадок отфильтровывают и экстрагируют из него продукт ацетоном. Получают 0,1 г (11%) 3,4-дийод-2-метил-5-формилпиррола(8). Rf= 0,8 (Г:ЭА 1:1); ПМР (CDCl3) d(м.д.): 2.46-с. (3Н; СН3), 9,35-с. (1Н; СНО), 10,13-уш.с. (1Н; NH). m/z(%) 361 (100%), 360 (60%), 235 (21%), 127 (47%).
2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазол.(9)
Вариант 1.
В колбе на 25 мл растерли 1 г (0,0028 моль) 2,5-диметил-3,4-дийодпиррола(5), 0,22 г (0,0028 моль) тиоацетамида. Смесь стояла в течение 10-15 мин при комнатной температуре. По данным ТСХ определили, что реакция закончена. Продукт отмыли бензолом и очищали с помощью колоночной хроматографии на силикагеле в системе растворителей Г : ЭА 8:1. Получили 0,15 г (32%) 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола(9) с Rf=0,6(Г: ЭА 8:1); ПМР (CDCl3) d(м.д.): 2,42-c. (3Н; CH3), , 2,48-с. (3Н; CH3), 2,58-с. (3Н; CH3), 7,94-уш. с. (1H; NH). m/z(%) 167(100%) [M+1].
Вариант 2.
В колбах на 25 мл приготовили растворы 0,5 г (0,0014 моль) 2,5-диметил-3,4-дийодпиррола(5) в 15 мл метанола и 0,11 г (0,0014 моль) тиоацетамида в 15 мл метанола. Далее оба раствора смешиваем в колбе на 50 мл и грели при 40°С в течении 5 мин. По данным ТСХ определили, что реакция закончена. Продукт очищали с помощью колоночной хроматографии на силикагеле в системе растворителей Г : ЭА 3:1. Получили 0,13 г (56%) 2,4,6-триметил-5Н-пирроло[3,4-d][1,3]тиазола(9) с Rf=0(Г : ЭА 3:1); ПМР (CDCl3) d(м.д.): 2,42-c. (3Н; CH3), , 2,48-с. (3Н; CH3), 2,58-с. (3Н; CH3), 7,94-уш. с. (1H; NH). m/z(%) 167(100%) [M+1].
4,6-диметил-5Н-пирроло[3,4-d][1,3]-2-аминотиазол.(10)В колбе на 25 мл растерли 0,5 г (0,0014 моль) 2,5-диметил-3,4-дийодпиррола(5), 0,11 г (0,0014 моль) тиомочевины, 0,1 г (0,0007 моль) K2CO3 и добавили 0,2 мл водного раствора метанола (1:1) и оставили стоять в течении 20 мин. По данным ТСХ определили, что реакция закончена. Продукт отмыли четыреххлористым углеродом и очищали с помощью колоночной хроматографии на силикагеле в системе растворителей ПЭ: ЭА 4:1. Получили 0,1 г (43%) 4,6-диметил-5Н-пирроло[3,4-d][1,3]-2-аминотиазола(10) с Rf = 0,8(ПЭ : ЭА 6:1); ПМР (CDCl3) d(м.д.): 2,22-с. (3H; CH3), 2,22-с. (3H; CH3), 5,87-д. (2H; NH2), 7,7-уш. с. (1H; NH).
Реакция 2,5-диметил-3,4-дийодпиррола с ортофенилендиамином.(11)
В колбе на 25 мл растерли 0,5 г (0,0014 моль) 2,5-диметил-3,4-дийодпиррола(5), 0,15 г (0,0014 моль) ортофенилендиамина, 0,1 г (0,0007 моль) K2CO3 и добавили 10 мл водного раствора метанола (1:1). Смесь стояла при комнатной температуре 40 мин, после чего по данным ТСХ определили, что реакция закончена. Продукт отмыли четыреххлористым углеродом и очищали с помощью колоночной хроматографии на силикагеле в системе растворителей ПЭ: ЭА 4:1. Получили 0,07 г (25%) продукта(11) с Rf = 0,8(ПЭ : ЭА 6:1). ПМР (CDCl3) d(м.д.): 2,19м.д. (3H; CH3), 2,3м.д. (3H; CH3), 5,66м.д. (1H; NH).
Похожие работы
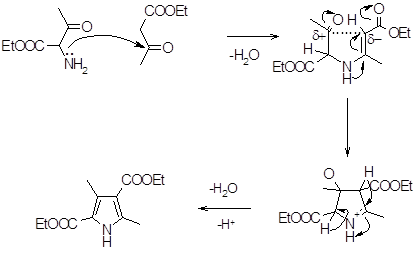



... техники безопасности, заботой об улучшении и оздоровлении условий труда, что очень актуально в настоящее время. Рассматриваемая магистерская диссертация, посвященная синтезу пиррольных интермедиатов для высоко сопряженных порфиринов, выполнена на кафедре ХТТОС МГАТХТ им. М.В.Ломоносова. Ниже анализируются токсичные и пожароопасные свойства веществ, использованных в работе, а также условия, ...
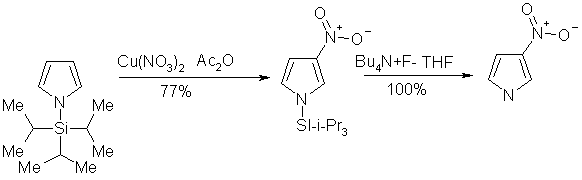
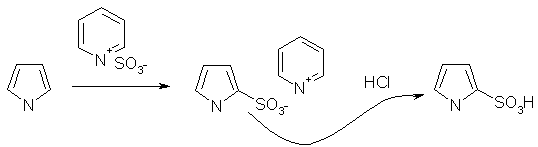
... продуктами олигомеризации и полимеризации. Наиболее гладко пиррол моноалкилируется сопряженными енонами, содержащими уходящую группу в β-положении. 1.2.2 Электрофильное замещение тиофена Как и следовало ожидать, для тиофена характерны реакции, свойственные ароматическим соединениям типа бензола. Тиофен галогенируется, нитруется, сульфируется и ацилируется аналогично фурану и пирролу ...

... комнатной температуре на несколько часов. Растворитель отгоняют в вакууме, а твердый остаток кристаллизуют из уксусной кислоты. Выход продукта 9-5%; т. кип. >360°. 2. Литературный обзор. Бензимидазол, его производные, их синтезы и свойства Данная работа посвящена изучению обладающих ценными свойствами бензимидазолов общей формулы их таутомеров, их стереоизомеров, их смесей ...
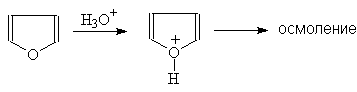
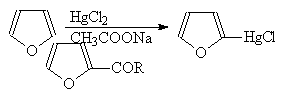
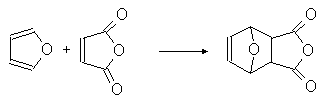
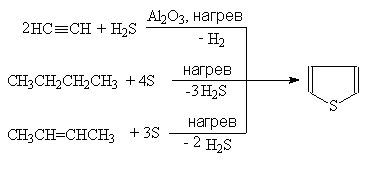
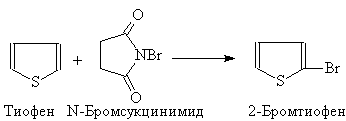
... гетероциклов. 2.1. Получение В промышленности тиофен производят по реакции ацетилена с сероводородом при 400оС или из н-бутана и серы в газовой фазе: (37) 2.2. Свойства Тиофен, в отличие от фурана и пиррола, не дает с обычными кислотами тиониевых солей и, следовательно, в кислой среде не утрачивает ароматических свойств. Он не обладает ацидофобностью. Тиофен легко ...
0 комментариев