Реферат на тему:
«Роль воспаления в повреждении миокарда при ишемии и реперфузии»
Природа повреждения миокарда при ишемии и реперфузии после достаточно длительного периода нарушенного кровоснабжения имеет много общих черт с воспалительным процессом, и традиционная противовоспалительная терапия способна оказывать в этих условиях защитное действие.
На изолированном сердце крысы показано, что реперфузируемый миокард характеризуется всеми признаками воспаления: высоким уровнем продукции свободных радикалов, усиленной инфильтрацией нейтрофилов, определяемой как гистологической, так и по миелопероксидазной активностью, возрастанием экспрессии мРНК КР-кВ.
Выраженность воспаления играет решающую роль в судьбе ишемизированного миокарда, и в условиях эксперимента блокада фактора КР-кВ при перевязке венечной артерии предупреждала развитие ИМ. Этот эффект был связан с уменьшением экспрессии генов ИЛ-6 и УСАМ-1, ослаблением инфильтрации миокарда моноцитами, нейтрофилами и лимфоцитами. Установлено, что защитное действие статинов в условиях реперфузии миокарда также объясняется их способностью инактивировать фактор NР-кВ. Это сочетается с ослаблением экспрессии МСР-1, ЦОГ-2 и ИЛ-8 [109, 203].
Реперфузионное повреждение миокарда связано с участием нейтрофилов, и между тяжестью поражения миокарда и активностью в нем специфичного для нейтрофилов фермента МРО существует прямая зависимость. Показано, что нейтрофилы накапливаются в зоне риска, активируются и высвобождают большое количество свободных радикалов кислорода, цитокинов, других провоспалительных медиаторов, протеолитических факторов, оказывающих цитотоксическое действие.
Участие воспаления в реперфузионном повреждении миокарда подтверждается патогенетической общностью этих процессов. В условиях реперфузии, как и при воздействии радикалов кислотов ЛПС и цитокинов типа ФНО-сс и ИЛ-1р, клетки эндотелия представляют экспрессию молекул адгезии Е-селектина, 1САМ-1, которую обусловливает ядерный фактор КР-кВ.
Общим звеном, приводящим к активации НР-кВ при реперфузии и при действии различных провоспалительных стимулов, является развитие оксидантного стресса в результате усиленной продукции активных форм кислорода, а увеличение внутриклеточного содержания восстановленных тиолов подавляет активацию НР-кВ, уменьшает выраженность повреждения как при первичном воспалении, так и в условиях реперфузии.
Усиленное образование при реперфузии противовоспалительных цитокинов, которое медиируется фактором КР-кВ, играет ведущую роль в активации эндотелия и интенсивной экспрессии 1САМ-1. При блокаде образования ФНО-а и предупреждении активации КР-кВ устраняются экспрессия 1САМ-1, адгезия и миграция нейтрофилов, уменьшается выраженность реперфузионного повреждения миокарда.
Оксидантный стресс является также одним из важнейших факторов артериального тромбоза и закупорки просвета микрососудов лейкоцитарными агрегатами, что лежит в основе невосстановления кровотока в условиях реперфузии.
Радикалы кислорода в течение нескольких секунд подавляют активность РАР-гидролазы, в результате чего возрастает содержание в крови РАР, что вызывает образование тромбоцитарных и лейкоцитарных агрегатов. Патогенетическая значимость радикальных форм кислорода в этом процессе подтверждается выраженным защитным эффектом антиоксидантов и скевенджеров свободных радикалов.
На протяжении первых 6 ч реперфузии происходит преимущественно внутрисосудистое накопление нейтрофилов, затем они совершают трансэндотелиальную миграцию и локализуются в интерстициальном компартменте.
Поэтому раннее реперфузионное повреждение определяется воздействием нейтрофилов на эндотелий и диффузией цитотоксических медиаторов к кардиомиоцитам с резким угнетением их сократительной функции, так называемой «оглушением миокарда». Более отсроченный эффект реперфузии, сочетающийся с гибелью миокардиальных клеток и обусловливающий расширение зоны некроза, сопряжен миграцией нейтрофилов через стенку сосуда и их непосредственным цитотоксическим действием на кардиомиоциты.
Подтверждением роли воспаления в реперфузионном повреждении миокарда является также возможность пропорционально уменьшения как интенсивности воспалительного ответа, выраженности некроза миокарда в условиях ишемии и реперфузии посредством активации РРАКу с помощью специфических агонистов.
Показано, что внутривенное введение розиглитазона в дозе 3 мг/кг на 37 % существенно уменьшало размер зоны некроза после окклюзии передней нисходящей ветви левой венечной артерии длительностью 30 мин с последующей 24-часовой реперфузией. Отмечали также уменьшенное на 43 % накопление нейтрофилов и моноцитов в зоне ишемии в сочетании с угнетением экспрессии на них молекул адгезии. Эти эффекты сочетались с более выраженным восстановлением сократительной функции миокарда [303].
Роль воспаления в развитии реперфузионных повреждений кардиомиоцитов проявляется также тем, что выраженность некроза миокарда и нарушений его сократительной функции четко коррелирует с интенсивностью высвобождения в перфузат ФНО-а, а применение антител к ФНО-а уменьшает тяжесть реперфузионного синдрома.
Этот эффект усиливается в гипертрофированном сердце, так как в нем продуцируется большее количество ФНО-а. Предварительное применение ингибитора также оказывает защитное действие и примерно в 4 раза улучшает постишемическое восстановление сократительной функции миокарда [287]. Так как между продукцией ФНО-а и индукцией 1МО8 существует прямая зависимость, это означает, что реперфузионное повреждение миокарда в значительной степени опосредовано усиленной продукцией N0 [268]. Реперфузионное повреждение миокарда в определяющей степени связано с экспрессией на эндотелии молекул адгезии 1САМ-1, последующей адгезией и трансэндотелиальной миграцией нейтрофилов.
Этот процесс, определяемый высвобождением ФНО-а и активацией фактора КР-кВ, протекает по классической двухэтапной схеме развития воспаления. На первом этапе происходит лабильный контакт нейтрофилов с эндотелиоцитами в виде их «прокатывания», которое определяется взаимодействием Р-сектина эндотелиоцитов с их лигандами (Р8ОЬ-1) на нейтрофилы и сопровождается экспрессией эндотелиоцитами молекул адгезии 1САМ-1.
В результате через 480 мин в реперфузии 1САМ-1-зависимая адгезия нейтрофилов возрастает в 3 раза по сравнению с контролем и в 2,5 раза по сравнению с после начала реперфузии. Устранение первичной адгезии лейкоцитов к эндотелию в первую фазу не оказывает существенного влияния на выраженность и динамику реперфузионного повреждения, тогда как предупреждение второго этапа адгезии сопровождается выраженным кардиопротекторным эффектом.
Роль лейкоцитов в развитии реперфузионных поражений была подтверждена и в исследованиях на мышах с генетическим отсутствием экспрессии молекул адгезии 1САМ-1. Показано, что через 2 ч реперфузии после 30 мин ишемии содержание в крови биохимических маркеров некроза (тропонина Т, КФК, ЛДГ) у мутантных мышей было значительно ниже, чем в контроле. Выраженность поражения, площадь некроза и количество инфильтрировавших лейкоцитов в зоне ишемии и в пограничной зоне также были уменьшены.
Однако размер рубца через 1 и 3 нед был сопоставимым в обеих группах мышей. Это означает, что защитный эффект блокады инфильтрации лейкоцитов в зону поврежденного миокарда характерен только для ранней стадии реперфузии, тогда как уже через 2 ч на фоне развившегося некроза лейкоциты играют существенную роль в фагоцитировании поврежденных клеток и «заживлении» некроза, способствуя образованию фиброзной ткани [173].
Повреждающее действие нейтрофилов, мигрировавших при реперфузии в зону ишемии, связано с их способностью высвобождать свободные радикалы кислорода (супероксидный и гидроксильный, перекись водорода), стимулировать ФЛ и усиливать гидролиз фосфолипидов клеточных мембран. В результате этого образуются РАР, который усиливает хемотаксис нейтрофилов, их адгезию к эндотелию, дегрануляцию и продукцию свободных радикалов. Кислородные радикалы продуцируются мембраносвязанным ферментом КАВРН-оксидазой, которая активируется находящимися в крови провоспалительными цитокинами, с компонентом комплемента, РАР, А II. Радикалы кислорода дополнительно стимулируют высвобождение из клеток крови и стенки сосуда провоспалительных медиаторов, что усиливает экспрессию гликопротеинового комплекса в нейтрофилах и молекулах адгезии 1САМ-1 на эндотелии в сочетании с угнетением продукции факторов, обладающих антиадгезивными свойствами (N0, аденозин, простациклин). РАР стимулируют также тромбоциты, которые действуют синергично с нейтролами.
Эти эффекты регулируются ауто-, пара- и эндокринным путем рядом провоспалительных медиаторов, высвобождаемых активированными нейтрофилами и эндотелиоцитами - ФНО-а, ИЛ-1Р, ИЛ-6, ИЛ-8, компонентами комплемента. Мигрировавшие в ткань активированные нейтрофилы дегранулируют с высвобождением протеаз, коллагеназ, ЛОГ, фосфолипаз, миелопероксидаз. Основным эффекторным механизмом повреждающего действия нейтрофилов является сериновая протеаза — эластаза, обусловливающая их деструктивный эффект на матриксные белки стенки сосудов.
Активированные нейтрофилы одновременно продуцируют СОР и N0, из которых образуется мощный цитотоксический оксидант ПОН, являющийся одним из важнейших медиаторов повреждения эндотелиоцитов. Этот эффект блокируется СОД, предупреждающей образование СОР, и в такой же степени угнетается донаторами N0 и стимуляторами еМОЗ, усиливающими продукцию N0 эндотелием. В то же время, в сочетанной культуре активация нейтрофилов вызывала гибель кардиомиоцитов, которая устранялась ингибиторами N0 и определялась, таким образом, цитотоксическим действием N0.
Это означает, что N0, продуцируемый в небольших количествах эндотелиальной еМО8, предупреждал повреждающее действие активированных нейтрофилов на эндотелиоциты, тогда как в высоких концентрациях N0, продуцируемый 1КО8 нейтрофилов, обусловливал их повреждающее действие на эндотелиоциты и кардиомиоциты, способствуя образованию ПОН [258].
Участие нейтрофилов в остром воспалительном ответе, инициированном ишемией миокарда, связано также с тем, что у лиц с гемодинамически значимым стенозом циркулирующие лейкоциты уже частично активированы, и их люминолусиленная хемилюминесценция увеличена более чем на 60 %. Поэтому количество лейкоцитов в крови и уровень их активности являются достоверным прогностическим признаком риска возникновениями тяжести его течения у пациентов со стабильной и нестабильной стенокардией [265].
Результаты исследований последних лет свидетельствуют о том, сколько существующие представления о механизмах взаимодействия клеток крови со стенкой сосудов еще далеки от реальных. Показано, что хемотаксис и прочная адгезия лейкоцитов, всего моноцитов и Т-лимфоцитов, медиируются также фракталкином — хемокином, который экспрессируется на эндотелиоцитах, активированных провоспалительными цитокинами М1Р-у и ФНО-а. Фракталкин — уникальный хемокин, который функционирует не только как хемоаттрактант, но и как молекула адгезии. Его рецептор (СХЗСКЛ) экспрессируется на макрофагах и эффекторных цитотоксических лимфоцитах, включая МК и цитотоксические Т-лимфоциты, которые содержат большое количество перфорина и гранзима. Растворенный в крови фракталкин вызывает миграцию этих клеток, тогда как фиксированный на мембране фракталкин обеспечивает их захват, усиленную миграцию в стенку сосуда и активацию с возрастанием цитотоксичности и усилением продукции 1Р1Ч-у.
В последние годы установлено, что фракталкин вовлекается в патогенез различных процессов типа атеросклероза, гломеру-лонефрита, ревматоидного артрита, отторжения трансплантата. Полиморфизм гена СХЗСКЛ, сочетающийся с его ослабленной экспрессией, существенно снижает риск развития острых коронарных событий, а при генетическом дефиците СХЗСКЛ у мышей с отсутствием отмечено на 50 % менее выраженное накопление макрофагов в стенке аорты, более значительное накопление ГМК и коллагена, что свидетельствовало о стабильном состоянии поражения [47].
Реперфузионное повреждение миокарда в значительной степени обусловлено активацией и миграцией в него нейтрофилов! под влиянием хемотаксических факторов, источником которых являются, прежде всего, кардиомиоциты, поврежденные в результате ишемии. Показано, что лимфа, оттекающая от ишемизированного миокарда, способна вызывать весь комплекс возникающих при реперфузии реакций с активацией нейтрофилов, экспрессией на них адгезией к сосудистому эндотелию. Этот процесс возникает сразу после начала реперфузии и сохраняется с уменьшающейся интенсивностью в течение более 4 ч.
Избирательность рекрутирования нейтрофилов в зону реперфузии обусловлена усиленной экспрессией в ней специфически для нейтрофилов хемоаттрактантов, КС (цитокин-индуцированного хемоаттракта нейтрофилов) и М1Р-2 (макрофагального воспалительного протеина-2). Экспрессия ЫХ характерна для кардиомиоцитов, индуцируется ФНО-а или свободными радикалами в условноксидантного стресса, блокируется ингибиторами КР-кВ, и ее нейтрализация на 80 % угнетает инфильтрацию нейтрофилов в ишемизированный миокард. В то же время, экспрессия КС и М1Р-2 осуществляется инфильтрировавшими нейтрофилами, и ее угнетение предупреждает рекрутирование нейтрофилов соответственно только на 28 и 37 %.
Одним из наиболее мощных факторов инициации острого воспаления при ишемии/реперфузии миокарда, приводящим к некрозу и апоптозу кардиомиоцитов, является ИЛ-113. При подавлении его действия с помощью трансфекции крысам гена блокатора рецепторов ИЛ-1 (ИЛ-Ка) 30-минутная окклюзия левой передней венечной артерии с последующей 24-часовой реперфузией сопровождалась развитием значительно меньшего (на 36 %) по размеру ИМ по сравнению с контролем; активность МРО в поврежденном миокарде, свидетельствующая об интенсивности инфильтрации лейкоцитов, была уменьшена на ТО %, а выраженность апоптоза кардиомиоцитов — на 50 % [260]. Показано также, что аденовирусная трансфекция гена ИЛ-Ка значительно ослабляет ответ на ишемию/реперфузию в мозге мышей [301].
Динамика процессов, возникающих в условиях тяжелой ишемии и некроза миокарда и лежащих в основе развития реперфузионного синдрома, ИМ и сердечной недостаточности, наиболее полно представлена в работе [82].
Прежде всего, некроз миокарда вызывает активацию комплемента и образование свободных радикалов, а также запускает цитокинный каскад посредством высвобождения ФНО-а и усиления экспрессии фактора КР-кВ. Активация системы комплемента при некрозе миокарда связана с прохождением через сарколемму кардиомиоцитов фрагментов митохондриальной мембраны, богатых кардиолипином и белками. Они связываются с компонентом комплемента, инициируют рекрутизацию моноцитов и нейтрофилов и запускают воспалительную реакцию. Поэтому лимфа, оттекающая от поврежденного миокарда, обладает хемотаксической активностью и вызывает
Репрессию ИЛ-6 в моноцитах. Этот эффект полностью угнетается нейтрализующими антителами к компоненту С5а комплемента. Усиленный синтез хемоаттрактанта ИЛ-8 и компонента С5а играет решающую роль в рекрутировании нейтротической резкой активации воспалительной реакции в реперфузируемом миокарде.
Похожие работы
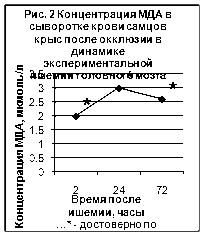

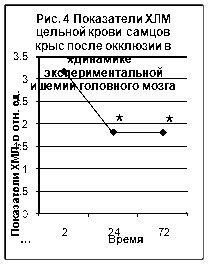

... ТБК-активных продуктов – малонового диальдегида (МДА), показателям люминолзависимой хемилюминесценции. 3.2. Исследование активности хемилюминесценции цельной крови и концентрации МДА в сыворотке крови при экспериментальной ишемии головного мозга Центральное место в изучении ишемического инсульта занимают экспериментальные модели на животных [65]. Именно по результатам эксперимента ...
... макрофагах, продуцирует в 100—1000 раз больше N0 по сравнению с постоянно экспрессированной еМО5. В малых концентрациях N0 является одним из важнейших физиологических регуляторов, тогда как в высоких он становится медиатором воспалительной реакции и оказывает выраженное цитоток-сическое действие. N0 быстро связывается с гемоглобином, который переводит его в неактивные продукты - нитриты и нитраты ...
... - в 14,7 раза, адгезия моноцитов к эндотелиальному монослою возросла на 48 % [163]. Одним из важнейших факторов, запускающим при воспалении трансэндотелиальную миграцию моноцитов, является активирующее действие нейтрофилов на клетки эндотелия. В результате этого стимулируется продукция и высвобождение эндоте-лиоцитами МСР-1 и повышается хемотаксическая активность моноцитов, что способствует их ...
... стабильным или бессимптомным течением ИБС. Это убедительно свидетельствует том, что воспаление, лежащее в основе дестабилизации ИБС, имеет самостоятельный характер и не зависит ни от выраженности стенозирующего поражения венечных сосудов, ни от некроза миокарда. Значимость системного воспаления в развитии ОКС была подтверждена также в большом проспективном исследовании с участием 15 тыс. здоровых ...
0 комментариев